
Le syndrome du QT long
Description
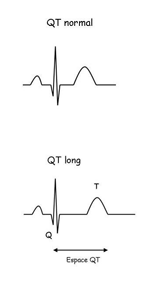
Le syndrome du QT Long (SQTL) est une maladie génétique rare du système électrique du cœur. Le système électrique du cœur permet la contraction du cœur. Chaque battement cardiaque résulte de la propagation dans les oreillettes puis dans les ventricules d’un ‘courant électrique’. Ce courant électrique engendre une ‘dépolarisation’ des cellules musculaires du cœur. En effet, les cellules musculaires sont comme des petites ‘batteries’, chargées électriquement. Cette charge électrique provient de l’existence sur leur paroi (membrane) externe de petits canaux ioniques. Le courant électrique engendre donc une dépolarisation électrique, responsable de la contraction des muscles cardiaques. Il en suit une ‘repolarisation’ spontanée des différentes cellules mettant donc le système de nouveau prêt pour une nouvelle contraction. C’est la dépolarisation et la repolarisation des oreillettes puis des ventricules qui sont enregistrées et mises sur papier par l’électrocardiogramme.
Dans le SQTL, il existe des anomalies des canaux ioniques présents dans les membranes cellulaires du cœur. Il en résulte des anomalies de la repolarisation électrique des ventricules. Ces défauts se caractérisent à l’électrocardiogramme par un l’allongement de l’intervalle QT, qui est le témoin de la repolarisation (figure). Cette anomalie de la repolarisation favorise la survenue chez les malades d’une forme typique d’accélération anormale du rythme cardiaque (arythmie ventriculaire) appelée torsade de pointe. Ces graves perturbations de rythme cardiaque entraînent une perte de conscience soudaine (syncope, pause cardiaque) et peuvent mener à la mort subite par fibrillation cardiaque. A noter que le cœur lui même n’est pas impliqué dans cette anomalie, sa fonction mécanique est normale.
Le SQTL atteint 1 personne pour 5000 dans le monde. Ce syndrome est responsable de nombreuses morts soudaines, chez les jeunes enfants, adolescents et jeunes adultes. D’après les statistiques américaines, 3000 décès par an seraient liés au syndrome du QT long chaque année dans ce pays (pour rappel la population des USA compte plus de 300 millions d’habitants).
Il existe deux formes ‘cliniques’ du SQTL
- Le SQTL de Jervell et Lange-Nielsen est rare. Les symptômes sont sévères et accompagnés de surdité dès la naissance. La transmission y est de type autosomique récessif ce qui veut dire que la maladie peut survenir chez les garçons et les filles (autosomique), mais que l’enfant doit avoir reçu un gène anormal de la mère ET du père pour être malade (récessif).
- Le SQTL de Romano-Ward est plus fréquent. Il n’y a pas de surdité associée. La transmission y est de type autosomique dominant ce qui veut dire que la maladie peut survenir chez les garçons et les filles (autosomique), mais qu’il suffit d’être porteur d’un seul gène, reçu donc soit de la mère , soit du père pour être malade. Par définition, le parent ayant transmis le gène est donc également ‘malade’. Dans cette forme, chaque enfant d’un parent affecté a donc une chance sur deux d’hériter du gène anormal et donc de la maladie.
Un ‘QT long’ peut également être acquis. Dans ces cas, l’allongement du QT est secondaire à une cause ‘extérieure’ et non inscrit dans les gènes. Les causes sont entre autres : un trouble ionique grave comme par exemple une hypocalcémie ou hypokaliémie (déficit en calcium ou en potassium dans le sang), une maladie sévère du système nerveux central ou plus fréquemment la prise de certains médicaments perturbant la repolarisation. Il va de soi que les patients porteurs d’un SQTL héréditaire doivent à tout prix éviter ces troubles ioniques ou ces médicaments allongeant l’intervalle QT, car ceux-ci peuvent précipiter les symptômes.
La génétique
Des progrès spectaculaires ont été faits dans le domaine de la génétique du SQTL. Depuis la découverte, en 1991, du 1er gène responsable du SQTL, d’autres gènes ont pu être identifiés. En effets, le SQTL peut être provoqué par des mutations (modifications) de gènes différents.
Par ailleurs, les mutations d’un même gène, peuvent être différentes d’une personne atteinte à l’autre. Ceci explique les symptômes différents et la gravité différente d’une personne à l’autre.
Les mutations des gènes produisent les anomalies des canaux ioniques, responsables des troubles de la repolarisation et donc des arythmies.
Jusqu’à présent (2010), 12 gènes anormaux impliqués ont été identifiés et on parle donc du LQT1 , LQT2 …jusqu’au LQT12 en fonction de l’ordre de découverte du gène responsable. Les présentations cliniques de ces différentes formes sont variables.
Symptômes
Les arythmies cardiaques favorisées par l’anomalie de la repolarisation sont responsable de syncopes, malaises graves ou même de mort subite. Même si ces malaises se produisent habituellement pendant l’effort ou juste après, ils peuvent également survenir durant une activité calme. Les événements déclencheurs potentiels sont nombreux tels les excitations, les émotions, la colère, la peur, les pleurs, le stress, les bruits stridents, une sonnerie de téléphone, le réveil du matin, un klaxon, des examens médicaux, des douleurs, etc. Ils varient en fonction du type génétique (LQT1 ou LQT2 etc.). Les vertiges peuvent également faire partie des symptômes.
La présentation clinique diffère d’une personne à l’autre, même au sein d’une même famille où tous les membres atteints présentent la même mutation de gène ! L’absence de symptômes ne signifie pas qu’une personne n’est pas atteinte. Environ 30 à 35 % des malades n’auront jamais de symptômes. Par ailleurs, le premier événement peut malheureusement être la mort subite. Les symptômes peuvent commencer dès le plus jeune âge mais aussi à l’adolescence, à l’âge adulte, lors d’un choc et pour les femmes lors d’une grossesse. Le sexe féminin semble être plus touché que le sexe masculin.
Diagnostic et prise en charge
Le diagnostic se fait par la mesure de l’intervalle QT sur l’électrocardiogramme. Cependant l’interprétation reste parfois difficile et délicate et 10% des patients, porteurs d’un gène anormal, ont un QT normal à l’électrocardiogramme (ECG).
Des tests génétiques, encore longs et coûteux à ce jour, doivent être orientés en fonction de la symptomatologie. Ils ne permettront malheureusement pas de trouver le gène anormal dans tous les cas.
Lorsqu’un diagnostic de SQTL est suspecté ou confirmé chez un membre d’une famille, il est très important que tous les membres et parents de cette famille se soumettent à des tests de recherche du SQTL (ECG + éventuellement tests génétiques).
Toutes les personnes atteintes du SQTL doivent être traitées, même si elles ne sont pas symptomatiques. Actuellement il n'est pas possible de guérir du SQTL, mais les différentes thérapies permettent d'améliorer l'état de santé des malades en réduisant le risque d’arythmies graves et en permettant ainsi une vie souvent très proche de la normale. Le choix du traitement est toujours en fonction des symptômes, des intolérances et des contre-indications que le malade peut avoir. Les médicaments les plus prescrits sont les bêtabloquants. Ils sont efficaces dans 85 à 90% des cas. Les bêtabloquants ont pour effet de bloquer l'action du système nerveux sympathique sur le cœur qui favorise la survenue de ces arythmies. Le propanolol (indéral®) et le nadolol (corgard®) sont les bêtabloquants les plus employés. L'association de bêtabloquants et la pose d'un pacemaker peut être envisagée si le traitement par bêtabloquants engendre une bradycardie (fréquence cardiaque trop lente) trop importante. L’implantation d’un défibrillateur ou autres traitements peuvent s’avérer nécessaire en cas de symptômes ne répondant pas aux bêtabloquants.
La natation, la course, tout comme les autres efforts physiques sont à proscrire ou du moins à pratiquer avec la plus grande attention. Il est important que les personnes atteintes du SQTL aient pris connaissance des médicaments pouvant prolonger le QT et strictement interdits chez eux. Il est donc important qu’ils puissent recevoir du cardiologue traitant une liste complète et claire. Les personnes atteintes du SQTL ne peuvent en aucun cas rester sans thérapie même pour une courte durée car il est impossible de prédire le risque de malaise, qui peut par ailleurs être fatal dès le premier épisode.
Liste des médicaments à éviter : liste
 retour Les principaux "syndromes" génétiques
retour Les principaux "syndromes" génétiques
Le syndrome de Marfan.
Description
Le syndrome de Marfan est une maladie génétique qui atteint les tissus conjonctifs. C’est le Dr Antoine Marfan, qui lui a donné son nom, en décrivant la maladie en 1896. Le tissu conjonctif est une partie importante des organes. En effet, il agit comme une colle et comme un échafaudage et donne ainsi structure, forme, force et élasticité aux différents organes tels les os, les muscles, les vaisseaux sanguins etc. Plusieurs protéines entrent dans la composition du tissu conjonctif, dont la fibrilline, l’élastine etc. Dans la maladie de Marfan, il y a une anomalie du gène fabriquant la fibrilline. Cette anomalie est responsable d’un affaiblissement des tissus conjonctifs. La fibrilline est codée par un grand gène (le FBN1) localisé sur le chromosome 15. C’est un gène de grande taille et plusieurs mutations différentes peuvent en conséquence y survenir et engendrer une maladie de Marfan. Un deuxième gène est impliqué (le MFS2) localisé sur le chromosome 3, qui rendrait compte d'environ 8 à 15% des syndromes de Marfan. D’autres gènes encore inconnus pourraient être impliqués. Actuellement, aucune corrélation entre la localisation d'une mutation dans le gène FBN1 et les signes cliniques n’a été retrouvé.
La maladie de Marfan affecte une personne sur 5000. Il s’agit d’une maladie autosomique dominante ce qui veut dire que la maladie peut survenir chez les garçons et les filles (autosomique), mais qu’il suffit d’être porteur d’un seul gène anormal, reçu donc soit de la mère, soit du père pour être malade (dominant). Par définition, le parent ayant transmis le gène anormal est donc également ‘malade’. Chaque enfant d’un parent affecté a donc une chance sur deux d’hériter du gène anormal et donc d’être ‘malade’. A noter cependant que dans 25-30% des cas, aucun des parents n’est atteint. Il s’agit alors d’une néo-mutation, ce qui veut dire que la modification du gène s’est produite dans l’œuf de la mère ou dans le spermatozoïde, d’un parent non atteint.
Symptômes et diagnostic
Chez les personnes atteintes du syndrome, l'affaiblissement du tissu conjonctif touche principalement le système cardio-vasculaire, le squelette, les ligaments et les yeux mais d’autres systèmes peuvent être atteints. Les symptômes squelettiques les plus fréquents sont la grande taille, la laxité des ligaments et une scoliose. Au niveau oculaire on note le plus souvent une myopie et un déplacement du cristallin, et au niveau cardiaque une dilatation de l’aorte et un prolapsus de la valvule mitrale.
Le diagnostic du syndrome de Marfan est un processus complexe qui demande l'intervention de plusieurs spécialistes. En effet, une personne ne présente jamais tous les signes de la maladie et certains signes sont aussi présents chez des personnes ne souffrant pas du syndrome. Le diagnostic nécessite l’association de plusieurs signes cliniques. L’histoire génétique ou familiale intervient également dans le diagnostic. Il est souvent difficile de poser le diagnostic sur base des signes cliniques avant l’âge de 5 ans. Une biopsie de la peau pour analyse de l’histologie (structure) de la peau et en particulier de la fibrilline peut être faite pour conforter le diagnostic clinique. Des tests génétiques permettent dans certaines familles d’identifier le gène présentant une mutation et ainsi d’identifier les autres membres de la famille atteints par le syndrome.
Atteinte cardiovasculaire
L’atteinte cardiovasculaire conditionne la sévérité du syndrome de Marfan puisque les complications peuvent mettre la vie en péril. Les principales atteintes possibles au niveau du cœur sont la dilatation de l’aorte surtout dans sa portion ascendante, le prolapsus mitral et la survenue d’arythmies.
La dilatation de l’aorte ascendante est l’atteinte qui mérite le plus d’attention. L’aorte reçoit à chaque contraction une grande quantité de sang à partir du ventricule gauche, sang qui est ensuite distribué aux différents organes. La valve aortique est située entre le ventricule gauche et l’aorte et empêche le sang de retomber dans le ventricule (valve anti-reflux). Avec le temps, suite à l’arrivée continue de sang dans l’aorte, celle-ci peut se dilater dans sa première partie (aorte ascendante). Chez le patient avec syndrome de Marfan, suite à la fragilité de la paroi, cette dilatation survient plus tôt et est plus sévère. Une dilatation peut être présente dès les premières années de vie ou même avant la naissance ou alors apparaître beaucoup plus tardivement. La simple dilatation de l’aorte ne donne aucun symptôme. Deux complications de cette dilatation sont cependant possibles : la fuite de la valve aortique et la dissection ou rupture de l’aorte.
La fuite de la valve aortique (insuffisance aortique) résulte directement de la dilatation de la racine aortique (aorte ascendante). Elle provoque une ‘régurgitation’ de sang vers le ventricule gauche, provoquant une surcharge de travail et une dilatation de ce ventricule. Ceci se manifeste par un essoufflement et une moindre tolérance à l’effort. Un patient avec une fuite aortique peu importante peut cependant être tout fait asymptomatique. A l’auscultation, un souffle est audible.
Une dilatation de l’aorte rapidement progressive ou existante depuis longtemps peut se compliquer d’une dissection de la paroi, complication très grave et pouvant être rapidement mortelle en absence de traitement. La dissection résulte d’une déchirure de la paroi interne de l’aorte. Ceci permet au sang de passer entre la paroi interne et externe et peut se solder par une rupture de l’aorte, souvent fatale. Une forte douleur à la poitrine ou dans le dos ou le cou peuvent être les signes d’une dissection, nécessitant une hospitalisation urgente. Cette douleur peut être plus sourde et s’accompagner de malaise, douleur ou mauvaise perfusion des membres, de symptômes évocateurs d’un accident vasculaire cérébral ou d’autres symptômes peu spécifiques. Le risque de dissection est proportionnel au degré de dilatation de l’aorte, survenant essentiellement lorsque le diamètre de l’aorte ascendante dépasse 55 mm (chez l’adulte). L’histoire familiale est également importante : en effet, si un membre de la famille a présenté une dissection à un diamètre moins important, le risque d’une histoire similaire pour un autre membre de la famille est plus important.
Le prolapsus mitral est fréquent en cas de maladie de Marfan. En raison du manque de ‘solidité’ des tissus conjonctifs, la valve mitrale et les cordages qui l’attachent au ventricule peuvent se distendre. Ceci peut favoriser une fuite de la valve mitrale (insuffisance mitrale). Une fuite importante de la valve mitrale engendre une moindre tolérance à l’effort et un essoufflement. Elle peut favoriser l’apparition d’arythmies, donnant une sensation de palpitations ou malaise. Un patient avec une fuite mitrale peu importante peut cependant être tout fait asymptomatique. A l’auscultation, un souffle est audible.
Les arythmies sous forme d’extrasystoles ventriculaires ou supra-ventriculaires semblent survenir un peu plus fréquemment chez le patient avec maladie de Marfan.
Le suivi cardiologique de tout patient avec maladie de Marfan impose la réalisation à intervalles réguliers d’échocardiographies avec mesures de la racine aortique, analyse de la valve aortique et mitrale. Chez l’enfant, les mesures de la racine aortique doivent être analysées en fonction de la surface corporelle. La surface corporelle se mesure sur base de la taille et du poids de l’enfant. Il existe des abaques, semblables aux courbes de croissance, permettant de déterminer si les dimensions mesurées sont normales pour la surface corporelle de l’enfant. En général, un examen échocardiographique est recommandé tous les 6 à 12 mois, et ce dès la petite enfance. La résonance magnétique et le scanner thoracique peuvent également mettre en évidence la dilatation aortique, avec l’avantage de mieux visualiser la crosse aortique dans toute son entièreté. Ce sont par contre des examens plus couteux et moins ‘faciles’ à réaliser que les échocardiographies, surtout chez l’enfant et ils ne sont donc pas proposés à la même fréquence que les échocardiographies. En cas de suspicion d’arythmies, un Holter ECG (enregistrement de 24 heures) et/ou une épreuve d’effort peuvent être réalisés.
Un groupe de médicaments permet de ralentir la progression de la dilatation de l’aorte : les bêtabloquants. Ils agissent sur le cœur et sur les vaisseaux avec comme effet de ralentir le cœur et de baisser la tension artérielle systolique. La pression sur la paroi aortique est donc moindre et survient moins souvent. Le traitement par bêtabloquants est conseillé dès le diagnostic de maladie de Marfan, même chez l’enfant. Des études récentes plaident pour l’efficacité des inhibiteurs des récepteurs de l’angiotensine 2 (sartans). Des études supplémentaires sont en cours chez l’enfant et chez l’adulte.
Lorsque la dilatation atteint un niveau où le risque de dissection devient trop important (> 55 mm ou parfois avant), une chirurgie de remplacement de la racine aortique est proposée. Si la valve aortique est très fuyante, il peut être nécessaire de remplacer la valve par une valve mécanique. Ces interventions sont heureusement rarement nécessaires pendant l’enfance. Une fuite mitrale importante peut également nécessiter une intervention chirurgicale.
La dissection aortique est une urgence médico-chirurgicale et nécessite immédiatement le transfert du patient dans une unité hospitalière à même de réaliser une intervention vasculaire. La dissection de l’aorte est exceptionnelle pendant la petite enfance mais peut survenir dans la deuxième décennie. Toute symptomatologie pouvant faire suspecter une dissection mérite donc une investigation urgente, même chez un jeune adolescent. Une échocardiographie transoesophagienne, une résonance magnétique et/ou un scanner thoracique sont les examens permettant de confirmer le diagnostic de dissection. En cas de confirmation du diagnostic, une intervention chirurgicale visant à remplacer la racine aortique est programmée sans délai.
Le sport n’est pas totalement déconseillé chez le sujet avec maladie de Marfan, cependant l’importance des lésions squelettiques et ligamentaires, oculaires et cardiaques peut contre-indiquer certaines activités. Ainsi la présence d’une dilatation de la racine aortique représente une contre-indication aux sports de contact (boxe, arts martiaux, hockey sur glace ou sur herbe …) ou autres sports nécessitant des efforts soutenus (équitation, squash, volleyball …). Il est important que chaque patient puisse discuter ces aspects très importants de façon individuelle avec les différents médecins impliqués. Un certain niveau d’activité physique doit être encouragé chez la plupart des patients tout en tenant compte de ‘ses particularités’.
Liste donnant quelques conseils en terme de sports chez le patients avec Syndrome de Marfan
 Les sports et le Marfan Les sports et le Marfan
Les 2 grands types de sport :
- Les exercices dynamiques (isocinétiques) s'accompagnent d'une modification de la longueur des muscles et d'un mouvement des articulations, mais la force intramusculaire générée est peu importante. La marche, la natation en sont des exemples. Au cours de ces efforts, la libération d'oxygène au niveau du muscle squelettique qui participe à l'exercice doit être maximale. Les vaisseaux périphériques se dilatent, si bien que la pression artérielle s'élève peu malgré l'augmentation du débit cardiaque. La pression appliquée sur la paroi aortique n'est donc que peu élevée.
- A l'opposé, les efforts statiques (isométriques) supposent une grande force avec peu de mouvement. L'haltérophilie en est l'archétype. Au cours de ces efforts très intenses de courte durée, la compression musculaire empêche la dilatation des vaisseaux qui sont dans le muscle. La pression artérielle augmente beaucoup, d'autant plus que la masse musculaire impliquée dans l'effort est importante, et que l'effort est intense. La pression appliquée sur la paroi aortique est donc très élevée, ce qui favorise la dilatation et la dissection aortiques.
La plupart des sports comprennent une composante dynamique et une composante statique, non seulement lors de la réalisation des matchs, mais également lors de l'entraînement. La classification des sports est donc un peu artificielle : l'importance du retentissement de l'effort dépend du niveau d'effort fourni auquel peut s'ajouter au cours d'une compétition l'émotion qui élève également la pression artérielle.
En cas de maladie de Marfan, les sports peuvent :
| - |
favoriser la dilatation aortique et sa dissection si on ne respecte pas les règles ci dessous, et ce d'autant plus qu'on ne prend pas de bêtabloquants (qui vont limiter l'augmentation de fréquence cardiaque et de pression artérielle au cours de l'effort). |
| - |
favoriser une luxation du cristallin, par les chocs, les accélérations et décélérations brutales. |
| - |
compliquer les problèmes orthopédiques |
Il est donc la règle de recommander :
| - |
de faire du sport sans esprit de compétition. |
| - |
de limiter les arrêts brutaux, les chocs avec les autres joueurs. |
| - |
de se limiter à 50% de la capacité maximale. |
| - |
d’éviter les sports isométriques. Porter plusieurs fois de petites charges est préférable à porter une fois une grosse charge. |
| - |
d’éviter de tester ses limites. |
Voici une liste des sports et leur degré de contre-indication:
Autorisés sans restriction :
Golf - Billard - Bowling - Cricket -Tir à la carabine - Tir à l'arc- Yoga - Taï chi.
Autorisés en dilettante :
Ping-pong - Marche - Footing - Bicyclette - Natation - Plongée - Equitation - Voile - Baseball - Volleyball - Danse.
Autorisés avec les enfants :
Football - Tennis en double - Jeux de volants.
Interdits :
Lever de poids - Musculation - Bobsleigh - Luge - lancer de marteau - Sports Martiaux - Escalade - Ski nautique - Planche à voile - Escrime - Saut en hauteur - Rodéo - Rugby - Sprint - Ski de descente - Squash - Tennis - Basket - Hockey - Boxe - Combat - Décathlon - Canoë - Kayak - Course automobile - Course de moto - Badminton - Gymnastique - Step - Reeboock - Athlétisme - Aviron.
En résumé
La maladie de Marfan s'accompagne d'une faiblesse de la paroi aortique qu'il faut éviter de soumettre à de trop fortes contraintes. Il est possible de pratiquer les sports d'endurance sans esprit de compétition.
Par contre, les sports de force, pratiqués en apnée, sont contre-indiqués (même en amateur) de même que les sports de combat. Le plus important est de comprendre la raison de cette limitation et de se l'appliquer soi-même, quelque soit le sport que l'on pratique.
|
retour Les principaux "syndromes" génétiques
Le syndrome de Williams.
Description
En 1961, le cardiologue néo-zélandais Williams décrit le cas de quatre enfants présentant des déficiences cardiaques congénitales, un visage caractéristique et un retard du développement mental. Un an plus tard, le cardiologue-pédiatre allemand Beuren décrivait quatre enfants très semblables. Le syndrome de ‘Williams-Beuren’ était né. La dénomination de ce syndrome est multiple : après "Syndrome de Williams-Beuren" on parle aujourd'hui de "Syndrome de Williams ". La littérature anglo-saxonne parle volontiers de "hypercalcémie infantile" tandis que la littérature plus ancienne, parle du "Syndrome de la face d'elfe".
Le Syndrome de Williams affecte 1 naissance sur 10.000, tant les garçons que les filles. Il résulte de la perte d’une petite partie du chromosome 7 (microdélétion 7q11.23). Cette perte de matériel chromosomique correspond à la perte d'un grand nombre de gènes, dont le gène responsable de la fabrication de l’élastine (ELN). L’élastine est un constituant important des tissus conjonctifs. Les tissus conjonctifs sont une partie importante des organes. En effet, ils agissent comme une colle et comme un échafaudage et donnent ainsi structure, forme, force et élasticité aux différents organes tels les os, les muscles, les vaisseaux sanguins etc. Plusieurs protéines entrent dans la composition du tissu conjonctif, comme l’élastine, la fibrilline. L’anomalie de l’élastine présente dans le syndrome de Williams interviendrait dans l’atteinte vasculaire et dans la dysmorphie (visage caractéristique). D’autres gènes, en partie identifiés, sont responsables des autres caractéristiques du syndrome. Ainsi, autre gène (LIMK1) serait impliqué dans les anomalies de repère dans l’espace que présentent ces enfants. La taille de la perte de gènes peut être variable d’un individu à l’autre ce qui pourrait expliquer la variabilité dans les formes cliniques.
La plupart des cas de syndrome de Williams sont sporadiques. Ceci veut dire que les parents sont en général sains. Dans ces cas là, le risque d’avoir un second enfant atteint du même syndrome est très rare. Le patient lui même a cependant une chance sur 2 de transmettre le syndrome à sa descendance.
Les signes cliniques et le diagnostic
Les symptômes possibles sont multiples bien qu’un enfant ne présente jamais l’ensemble des symptômes. Les symptômes les plus fréquents et marquants sont une dysmorphie du visage (trait particuliers du visage, anciennement repris sous le nom de ‘facies d’elfe’), une cardiopathie (voir ci-dessous), un retard mental avec un quotient intellectuel aux environs de 60, un comportement très spécifique caractérisé par une grande sociabilité et une sensibilité marquée au bruit ou à la musique et plus rarement un taux de calcium trop élevé dans le sang chez le nouveau-né (hypercalcémie).
L’association d’un certain nombre de signes cliniques permet d’évoquer le diagnostic avec une grande certitude. Le diagnostic final sera donné par le test génétique (caryotype avec recherche de la microdélétion par méthode ‘FISH’), qui retrouve la microdélétion dans 98% des cas.
Atteinte cardiaque
L’atteinte cardiaque est présente chez 70% des patients. La lésion la plus fréquemment rencontrée est la sténose supra-valvulaire aortique. Les sténoses des artères pulmonaires sont également fréquentes et peuvent être soit distales (périphériques) ou juste au dessus de la valve pulmonaire (supra-valvulaire pulmonaire). D’autres vaisseaux peuvent également présenter des rétrécissements (sténoses), en particulier les artères allant vers les bras et vers le cerveau (artères sous-clavières et carotides), les artères coronaires (perfusant le cœur) et les artères rénales (perfusant les reins). La valve mitrale peut, de façon beaucoup plus rare, être anormale. De façon générale, on constate des anomalies diffuses des vaisseaux artériels qui sont plus petits (hypoplasiques) et plus rigides que normalement. Ceci est le résultat de l’anomalie de l’élastine, décrite ci-dessus.
Le degré d’atteinte est très variable d’un enfant à l’autre et détermine la symptomatologie, l’évolution et le pronostic. La sténose supra-valvulaire aortique, la plus fréquente, peut s’aggraver dans le temps, devenir symptomatique et nécessiter une intervention chirurgicale. La présence des sténoses des artères coronaires associées peut majorer la symptomatologie. Les sténoses périphériques pulmonaires, quant à elles, sont plus rarement symptomatiques et s’améliorent souvent avec la croissance. Une intervention est donc plus rarement nécessaire. Une sténose d’artère rénale peut être à l’origine d’une hypertension artérielle. De façon générale ces enfants sont, même en l’absence d’une sténose de l’artère rénale plus à risque de développer une hypertension artérielle en raison de l’hypoplasie et de la rigidité de leurs vaisseaux artériels. L’hypertension artérielle, si elle est présente nécessite un traitement médical. Si l’hypertension résulte d’une sténose de l’artère rénale, un traitement par cathétérisme vasculaire ou par chirurgie peut être nécessaire pour lever l’obstacle. Les sténoses des autres vaisseaux (sous-clavières, carotides..) sont rarement symptomatiques.
Le suivi cardiologique de ces enfants nécessite donc essentiellement des examens cliniques réguliers avec mesure de la tension artérielle et échocardiographie. L’observation de sténoses des artères qu’elles soient pulmonaires, rénales ou autres peut néanmoins s’avérer difficile par simple échocardiographie et peut nécessiter d’autres techniques d’imagerie tel le scanner ou l’angiographie.
! ! Toute anesthésie chez un enfant présentant un syndrome de Williams, doit être réalisée par un anesthésiste connaissant le syndrome et ces caractéristiques. En effet les anomalies de l'élasticité des artères font que les patients ont plus de difficultés à maintenir des tensions artérielles adéquates lors d'une induction anesthésique avec malheureusement parfois la survenue d'hypotension sévère, de choc cardiogénique, voire de mort subite. Il est donc indispensable que les anesthésistes puissent adapter leur ‘technique’ d’endormissement à cette particularité en veillant au maintien d'une volémie (volume sanguin) et d'une tension artérielle normales.
retour Les principaux "syndromes" génétiques
Le syndrome de Noonan
Le syndrome de Noonan est décrit pour la première fois en 1963 par la cardiopédiatre Jaqueline Noonan. Il s’agit d’une affection génétique, touchant 1 enfant sur 2000 à 2500, tant filles que garçons et qui associe une petite taille, une cardiopathie congénitale et des traits particuliers du visage.
Le syndrome de Noonan est du à une altération d’un gène (mutation). Dans plus de 50% des cas, il s’agit d’une mutation du gène PTPN11 présent sur le chromosome 12. Un autre gène (K-RAS) a récemment été découvert comme étant responsable d’un petit nombre (5%) de syndromes de Noonan. En cas de suspicion clinique les recherches génétiques actuelles permettent de retrouver une mutation connue chez un peu plus d’1 patient sur 2. Si le test est négatif, c’est soit parce que le patient n’a pas le syndrome de Noonan soit parce que la mutation d’un autre gène est responsable du syndrome.
Le syndrome de Noonan est une maladie à hérédité autosomique dominante, ce qui veut dire que les enfants d’une personne atteinte (fille ou garçon) ont un risque de 50% d’hériter de la maladie. Il y a toutefois une prédominance de transmissions maternelles (c’est en général la mère qui est atteinte). Les cas sporadiques s'expliquent par des néo-mutations au niveau de l’œuf ou du spermatozoïde, chez un des parents par ailleurs sain.
Les signes cliniques et le diagnostic
L’expression et la gravité dépend fortement d’une famille à l’autre. Le syndrome de Noonan peut affecter à des degrés divers presque tous les organes et fonctions. Les signes les plus souvent retrouvés sont: des traits de visage particuliers, des anomalies cardiaques (voir ci-dessous), des difficultés alimentaires en bas âge, une petite taille, des difficultés d’apprentissage avec rarement cependant un vrai retard mental, des troubles auditifs, des anomalies hématologiques et lymphatiques.
Les manifestations cliniques ‘typiques’ permettent généralement de poser un diagnostic mais ceci peut être difficile. La génétique permet à l’heure actuelle de confirmer le diagnostic dans un peu plus d’1 cas sur 2.
Atteinte cardiaque
Une atteinte cardiaque est présente à la naissance chez 80% des enfants. Les lésions typiques sont la sténose valvulaire pulmonaire (fréquente) et la cardiomyopathie hypertrophique du ventricule gauche (plus rare).
En cas de sténose valvulaire pulmonaire, la valve pulmonaire est souvent plus épaisse et anormale (dysplasie) que dans le cas d’une sténose valvulaire pulmonaire chez un patient n’ayant pas le syndrome de Noonan. Les symptômes, l’évolution et le traitement sont les mêmes qu’en cas de sténose valvulaire pulmonaire ‘sans’ syndrome de Noonan. Cependant, le traitement par cathétérisme cardiaque (dilatation de la valve) s’avère souvent moins efficace qu’en l’absence de syndrome en raison de l’épaisseur anormale de la valve. Il faut donc plus souvent recourir à la chirurgie pour lever l’obstacle.
La cardiomyopathie hypertrophique du ventricule gauche est présente chez 20 à 30% des patients avec syndrome de Noonan. Elle peut être présente dès la naissance ou se constituer plus tardivement. Elle donne rarement des symptômes et nécessite donc rarement un traitement. Le suivi est fait par échocardiographie.
La réalisation d’un électrocardiogramme chez un patient avec syndrome de Noonan permet de parfois constater une anomalie de l’axe électrique du cœur (axe de l’onde QRS dirigé vers le haut), ce qui n’a aucune répercussion clinique et ne nécessite donc ni traitement ni suivi particulier.
retour Les principaux "syndromes" génétiques
Le syndrome de Turner
Le syndrome de Turner est une anomalie chromosomique entrainant chez la femme une petite taille et un mauvais fonctionnement des ovaires. De façon plus variable, il peut y avoir d’autres anomalies associées.
Le syndrome de Turner touche 1 femme sur 2500 à la naissance. Il est du à l’absence partielle ou complète d’un des chromosomes X. Pour rappel, la femme a normalement 2 chromosomes X (XX) par cellule alors que l’homme a normalement 1 chromosome X et 1 chromosome Y (XY). Dans environ la moitié des cas de syndrome de Turner, le chromosome X manque en entier. Dans plus de 20% des cas, le syndrome de Turner est en mosaïque, c'est-à-dire qu'il existe à la fois des cellules anormales (un seul chromosome X) et des cellules normales (deux chromosomes X) dans le corps. Enfin, dans 30% des cas environ, il existe 2 chromosomes X mais l'un des 2 est altéré dans sa structure. Ces anomalies sont accidentelles.
Les signes cliniques et le diagnostic
Les manifestations cliniques sont très variables d’une personne à l’autre. La petite taille, présente chez 98% des patientes, peut être le seul signe pendant l’enfance. Une absence de développement pubertaire avec infertilité résultant d’une dysgénésie gonadique est très fréquemment rencontrée. Les autres anomalies moins fréquentes sont par exemple des anomalies du cœur (voir ci-dessous) et des reins, des particularités au niveau du visage et des membres, des problèmes de la thyroïde ou des otites à répétition. Un œdème (gonflement) des mains et des pieds peut être présent à la naissance associé à un cou palmé caractéristique. Le diagnostic est parfois suspecté avant la naissance par l’existence d’un œdème du cou ou œdème généralisé. Le développement intellectuel est en général normal. Les traitements hormonaux actuellement disponibles et bien codifiés pour ces patients-là ont grandement amélioré le devenir de ces patientes.
Atteinte cardiaque
Selon les études, 17 à 45% des patientes avec syndrome de Turner présentent une anomalie cardiaque. Celles-ci concernent essentiellement le cœur gauche : bicuspidie de la valve aortique, coarctation, hypoplasie du cœur gauche, anomalie du retour veineux pulmonaire. En fonction du type d’anomalie la présentation clinique peut être plus ou moins grave et se manifester à la naissance ou plus tard. Lors du diagnostic de syndrome de Turner, un bilan cardiaque par échocardiographie s’impose afin de dépister une éventuelle malformation cardiaque.
Il est actuellement reconnu que les patientes avec syndrome de Turner présentent une fragilité de la paroi aortique qui peut être responsable d’une dilatation de la racine aortique allant parfois jusqu’à la dissection aortique, comparable à celle des patients avec maladie des tissus conjonctifs comme le syndrome de Marfan, bien que l’atteinte soit moins fréquente et moins sévère dans le syndrome de Turner. Des contrôles échocardiographiques réguliers (1 fois par 3 à 5 ans en absence de dilatation, 1 fois par 6 à 12 mois en présence de dilatation) sont recommandés.
retour Les principaux "syndromes" génétiques
La microdélétion 22q11
Dès 1965, le Docteur DI GEORGE décrit pour la première fois les symptômes observés chez quatre enfants à la naissance : anomalie cardiaque associée à une anomalie de thymus et une hypocalcémie par anomalie des parathyroïdes. En 1978, le Docteur SHPRINTZEN décrit un aspect particulier du visage associé à une fente palatine, une anomalie du larynx et des troubles de l'apprentissage. Ce syndrome porte le nom de syndrome Vélo-Cardio-Facial ou VCFS ou encore de syndrome de Shprintzen. Dans les suites, les études génétiques montreront que le VCFS, le syndrome de Di George, mais également le syndrome cardiofacial de Cayler, le syndrome des anomalies conotroncales et de la face (décrit au Japon) sont à regrouper car tous liés à une microdélétion du chromosome 22. L’utilisation du terme ‘microdélétion 22q11’ est actuellement conseillée pour parler de ce syndrome. Le terme CATCH22, acronyme élaboré dans le monde anglo-saxon, reprenant les malformations les plus fréquentes du syndrome, doit être rayé de la liste des appellations.
La fréquence de la microdélétion 22q11 dans la population est estimée à 1/5000 naissances. La microdélétion 22q11 se transmet selon un mode autosomique dominant, ce qui veut dire qu’un parent atteint a une chance sur 2 de le transmettre à son enfant, garçon ou fille. Ce n’est cependant que dans 10 à 20% des cas que l'un des deux parents est trouvé porteur de cette microdélétion. L’accident chromosomique survient donc généralement de novo, c'est à dire de manière accidentelle (néomutation). Dans la microdélétion 22q11 il y a une délétion, c'est à dire une absence de matériel chromosomique au niveau de la bande q11 d'un des deux chromosomes 22. Plusieurs gènes de la région sont connus mais le ou les gène(s) responsables des anomalies observées n'ont pas encore été identifiés. Le gène TBX1, situé dans la région délétée pourrait être impliqué dans le phénotype cardiaque car son inactivation chez la souris est à l'origine de malformations cardiaques de type conotroncal, comme rencontré dans ce syndrome (voir plus bas)
Les signes cliniques et le diagnostic
Les symptômes possibles sont multiples bien qu’un enfant ne présente jamais l’ensemble des symptômes. Les symptômes les plus fréquents et marquants sont les anomalies du palais et des oreilles, des troubles de l’alimentation liés aux anomalies de la gorge ou du larynx, les malformations cardiaques, l’hypocalcémie en période néonatale, les troubles des défenses immunitaires, les malformations du système urinaire, la constipation, la scoliose, les troubles moteurs et difficultés spatio-temporelles, l’hypotonie, le retard dans les apprentissages et le déficit d’attention, les troubles psychiatriques chez l’adulte.
La microdélétion 22q11 est de description récente. Il existe actuellement encore peu de données précises sur sa fréquence exacte dans la population, le pourcentage de formes asymptomatiques ou peu sévères et le pronostic mental à long terme de ces enfants. La meilleure connaissance actuelle du syndrome permet cependant de mettre en place une prise en charge plus précoce, plus complète et plus globale des différentes facettes du syndrome et d’espérer ainsi un développement plus harmonieux des enfants.
La recherche de la microdélétion 22q11 doit être systématique chez tout enfant présentant un tableau clinique suggestif. La microdélétion 22q11 n'est pas visible sur un caryotype standard mais nécessite une technique spécifique (technique d'hybridation in situ ou FISH) à l'aide de sondes moléculaires testant la région 22q11. La recherche est positive chez plus de 95% des patients. Il est indispensable d'effectuer la recherche de la microdélétion chez les deux parents de tout enfant atteint. Lorsqu'aucun des deux parents ne présente la délétion, le risque de récidive lors d'une grossesse suivante est très faible. Un individu porteur de la microdélétion a un risque sur deux de la transmettre à sa descendance. Il est possible de rechercher la microdélétion sur des cellules amniotiques par hybridation in situ ou en utilisant les techniques de biologie moléculaire. Ce diagnostic prénatal peut être proposé lorsqu'un des deux parents présente la microdélétion ou encore devant la découverte d'une malformation évocatrice à l'échographie prénatale (cardiopathie congénitale). Il est cependant toujours très difficile de prédire l’importance et la gravité du tableau clinique.
Atteinte cardiaque
A peu près 75% des enfants avec microdélétion 22q11 présentent une malformation cardiaque à la naissance. Ceci s’explique par le fait que l’anomalie génétique perturbe pendant la formation de l’embryon, la migration vers la région du cœur de certaines cellules (crête neurale). Ces cellules doivent normalement, pendant la 4ième semaine de gestation, migrer vers les structures appelées les arcs branchiaux III et IV. Ces arcs branchiaux participent à la formation du cœur et des vaisseaux, en particulier à la formation de l’arc aortique et de la région ‘conotroncale’, qui anatomiquement correspond aux issues artérielles du cœur (figure). On a pu montrer chez l'animal qu'un blocage de cette migration provoquait des malformations dans cette région du cœur : c’est pourquoi dans la microdélétion 22q11, l’anatomie du cœur proprement dit est peu perturbée, au contraire de celle des grandes artères (aorte et artère pulmonaire) et de leur jonction avec les ventricules.
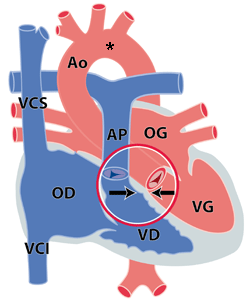
Figure : Cœur normal. Le cercle montre la région ‘conotroncale’ avec le ‘septum conal’ (entre les flèches). L’étoile montre la crosse de l’aorte. OD = oreillette droite, OG = oreillette gauche, VD = ventricule droit, VG = ventricule gauche, AP = artère pulmonaire, Ao = aorte
Les malformations les plus fréquemment retrouvées sont donc :
- les anomalies ‘conotroncales’ : la tétralogie de Fallot (17%), l’atrésie pulmonaire avec communication interventriculaire (CIV) (10%), le tronc artériel commun (9%), l’interruption de la crosse de l’aorte (14%), la CIV avec malalignement du septum conal (14%)
- les anomalies de la crosse aortique: le double arc aortique, la crosse aortique droite. ..
D’autres malformations sont occasionnellement rencontrées.
La présentation clinique et la prise en charge de ces différentes malformations sont évidemment très variables (voir chapitres correspondants). Certaines malformations sont très symptomatiques dès la naissance et nécessitent une prise en charge immédiate (comme par exemple l’atrésie pulmonaire avec CIV) alors que d’autres peuvent rester asymptomatiques toute la vie et ne nécessiter aucun traitement (par exemple crosse aortique droite).
Association belge de la microdélétion 22q11 (francophone) : Relais 22, www.relais22.be
 |
Association belge de la microdélétion 22q11 (francophone) : Relais 22 , www.relais22.be |
retour Les principaux "syndromes" génétiques
La trisomie 21
La trisomie 21 ou syndrome de Down est une maladie chromosomique congénitale provoquée par la présence d'un chromosome surnuméraire pour la 21e paire. Ses signes cliniques sont très nets : on observe un retard mental, associé à des modifications morphologiques particulières. La maladie a été décrite par le médecin britannique John Langdon Down en 1866. Il est l'auteur des mots ‘mongolien’ et ‘mongolisme’, proposés en référence aux yeux bridés des sujets atteints par le syndrome mais l'emploi de ces termes est aujourd'hui officiellement déconseillé.
La trisomie 21 est présente chez 1 enfant sur 1000 à la naissance. La présence d'un troisième chromosome 21 est la cause de la pathologie. Le mécanisme de la présence du chromosome supplémentaire (trisomie libre ou trisomie par translocation) est important à connaître pour le conseil génétique. La réalisation du caryotype permet de connaître ce mécanisme. L’âge maternel augmentant le risque de trisomie 21, un diagnostic anténatal par amniocentèse peut être proposé en fonction de ce risque. Une femme trisomique 21 a une probabilité de 50% d'avoir un enfant atteint.
Les signes cliniques et le diagnostic
Les manifestations cliniques sont très variables d’une personne à l’autre. La petite taille, les membres courts et le facies sont toutefois caractéristiques. La langue épaisse, associée assez souvent à une mâchoire étroite, entraînent surtout chez l'enfant à la sortir fréquemment de la bouche (protrusion de la langue). Les mains comportent un seul pli palmaire marqué et les doigts sont courts. Les anomalies musculo-squelettiques (hypotonie musculaire et hyperlaxité ligamentaire) sont constantes et peuvent entrainer des complications de type scoliose, luxations, instabilité articulaire, etc. Les anomalies cardiaques, digestives, rénales sont fréquentes et doivent être recherchées. Le retard mental est constant, bien qu’il puisse être variable d’un sujet à l’autre. La puberté est normale chez les filles, mais les garçons restent stériles. Les femmes ont une sexualité exacerbée. Le vieillissement est accéléré. Une incidence élevée de leucémie est constatée.
L’atteinte cardiaque
Près de la moitié des enfants avec trisomie 21 présentent une anomalie cardiaque. Celle-ci est le plus souvent de type ‘canal atrio-ventriculaire‘. D’autres anomalies sont possible comme le canal artériel persistant, la communication interventriculaire, la communication interauriculaire, la tétralogie de Fallot ou des associations par ailleurs plus rares comme l’association d’un canal atrio-ventriculaire et d’une tétralogie de Fallot ou l’association d’un canal atrio-ventriculaire et d’une coarctation.
La présentation clinique et la prise en charge de ces différentes malformations sont évidemment très variables (voir chapitres correspondants). Un traitement peut être proposé pour ces différentes malformations et permet dans la grande majorité des cas une ‘correction’ des anomalies, réduisant ainsi le handicap de ces enfants.
 |
Association belge de la trisomie 21 (francophone) : APEM-T21 , www.apem-t21.be |
retour Les principaux "syndromes" génétiques
Le syndrome d’Alagille
Le syndrome d’Alagille est défini par l'association de cinq éléments:
une cholestase (forme de ‘jaunisse’) chronique liée à une atteinte des voies biliaires dans le foie appelée ‘paucité’ des voies biliaires
une atteinte cardiaque, consistant en général en une sténose périphérique des branches de l'artère pulmonaire, mais une tétralogie de Fallot est également possible
des vertèbres « en ailes de papillon »
un visage caractéristique
une anomalie de l’œil (embryotoxon postérieur)
Seules l’atteinte des voies biliaires et la cardiopathie sont symptomatiques. La majorité des patients présentent comme premier signe une cholestase. Le syndrome peut toutefois se révéler sous la forme d'une tétralogie de Fallot. La recherche des anomalies oculaires et vertébrales permettra de confirmer le diagnostic.
Le syndrome d’Alagille atteint 1/100 000 naissances. La transmission est autosomique dominante, ce qui veut dire qu’un parent atteint à un risque de 50% de le transmettre à sa descendance, aussi bien les filles que les garçons. L’expressivité est variable autrement dit, d’une personne à l’autre, même à l’intérieur de la famille, la sévérité de la maladie peut être très différente. De nombreux cas sont sporadiques, impliquant que la mutation du gène est survenue sur l’œuf ou sur le spermatozoïde de parents ‘sains’. Des mutations du gène Jagged1, sur le chromosome 20 sont retrouvées chez 50 à 60% des malades.
La prise en charge de ces enfants implique le pédiatre hépatologue et le cardiologue pédiatrique. Au niveau hépatique, l’indication d'une transplantation hépatique est posée dans 40% des cas. Au niveau cardiaque, la ‘simple’ sténose périphérique des artères pulmonaires ne nécessite en général aucun traitement particulier mais elle peut néanmoins compliquer la prise en charge hépatique.
retour Les principaux "syndromes" génétiques
Le syndrome de CHARGE
Le mot CHARGE est un acronyme pour Colobome (anomalie de l’œil), malformation cardiaque (Heart), Atrésie des choanes (nez), Retard de croissance et/ou retard mental, hypoplasie Génitale et anomalies des oreilles et/ou surdité (Ear). Les enfants présentent donc un ensemble d'anomalies survenant de manière concomitante, plus fréquemment que si c'était le simple fait du hasard.
Les anomalies cardiaques sont présentes chez 75-80% des sujets. La cardiopathie la plus fréquente est la tétralogie de Fallot. Les autres anomalies sont la persistance du canal artériel, le ventricule droit à double issue éventuellement avec canal atrio-ventriculaire, la communication inter-ventriculaire ou inter-auriculaire secundum ou primum avec fente mitrale. D’autres malformations plus complexes peuvent également être rencontrées.
L’incidence réelle du syndrome est inconnue, mais les estimations vont de 1 cas pour 8 500 à 1 cas pour un million de naissances vivantes. Récemment, chez des patients présentant le syndrome, des mutations dans le gène CHD7, grand gène essentiel dans le développement embryonnaire du cœur, de l'oreille interne et de la rétine, ont été retrouvées.
La prise en charge des enfants avec syndrome CHARGE doit être intensive et multidisciplinaire car ces enfants requièrent fréquemment de nombreuses opérations chirurgicales ainsi qu’un suivi cardiologique, ophtalmologique, ORL et développemental.
retour Les principaux "syndromes" génétiques
L’association VACTERL
Le mot VACTERL est un acronyme pour des termes anglais signifiant malformations vertébrales (Vertebral defects), imperforation anale (Anal atresia), anomalies cardiaques (Cardiac), fistule trachéo-oesophagienne (TracheoEsophageal fistula) et anomalies du rein et des membres (Renal et Limbs). Les enfants présentent donc un ensemble d'anomalies survenant de manière concomitante, plus fréquemment que si c'était le simple fait du hasard.
Les malformations cardiaques les plus fréquentes sont la communication interventriculaire et interauriculaire et la tétralogie de Fallot. Le tronc artériel commun et la transposition des grands vaisseaux peuvent également être rencontrés mais moins fréquemment.
Le VACTERL et le VATER (sans atteinte cardiaque) affectent 1 cas pour 3333 à 6250 naissances. Presque tous les cas sont sporadiques, et aucune étiologie tératogène ou chromosomique n'est retenue. Une étiologie génétique n'a été rapportée que pour des cas de VACTERL avec hydrocéphalie.
Le traitement doit évidemment être pluridisciplinaire et comprendre la prise en charge des anomalies cardiaques.
retour Les principaux "syndromes" génétiques






