 Description
Description
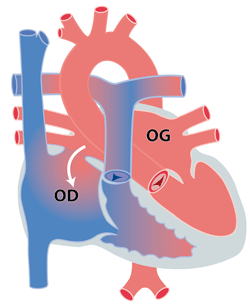
Le terme ‘communication interauriculaire’ ou CIA indique un passage existant entre l’oreillette droite et l’oreillette gauche (figure). Cette communication permet au sang rouge de passer de l’oreillette gauche (OG) vers l’oreillette droite (OD) et ainsi vers le ventricule droit (VD) puis les artères pulmonaires (AP). Ceci entraîne une augmentation du travail du cœur et une augmentation de sang dans les poumons.
Il faut néanmoins se rappeler qu’avant la naissance une communication existe et est indispensable entre les deux oreillettes. Il s’agit du foramen ovale ou trou de Botale. Celui-ci se ferme normalement dans les semaines ou mois qui suivent la naissance. Il reste cependant ouvert chez 1 personne sur 4 (‘FOP’ = foramen ovale persistant) mais dans ces cas le passage de sang est infime et sans conséquences pour le cœur.
La CIA peut être isolée ou associée à d’autres malformations importantes. Nous ne parlerons dans ce paragraphe que de la CIA isolée ou se présentant comme la malformation principale.
La CIA peut se localiser à différents endroits et il est très important de distinguer ces différentes localisations car la prise en charge médicale est très différente. On distingue ainsi la CIA secundum, situé dans la partie centrale de la cloison, la CIA primum située près des valves mitrales et tricuspides, et la CIA sinus venosus située près de la veine cave inférieure ou supérieure. Alors que la CIA secundum est souvent isolée, la CIA primum s’accompagne généralement d’une malformation de la valve mitrale (fente mitrale). La CIA de type sinus venosus, s’accompagne souvent d’anomalies d’implantation des veines pulmonaires droites.
La CIA isolée représente à peu près 7% des malformations cardiaques congénitales. Elle est plus fréquemment retrouvée chez les femmes.
Présentation clinique
Les enfants porteurs d’une CIA isolée, sont en général asymptomatiques à la naissance et pendant les premiers mois de vie. Lorsque la CIA est large on peut voir apparaître après quelques mois de vie, une difficulté de prise de poids, un certain essoufflement à l’effort et des infections respiratoires fréquentes. Chez la plupart des enfants cependant, ces symptômes sont peu prononcés voir même absents. C’est le plus souvent la présence d’un souffle ou la découverte d’une cardiomégalie (gros cœur) à la radiographie du thorax qui amène au diagnostic. Si cependant la CIA n’est pas traitée, les patients peuvent à l’âge adulte présenter des signes de défaillance du cœur droit (essoufflement à l’effort, gonflements des jambes..) ou des troubles du rythme (palpitations, tachycardie..).
Certaines CIAs, en fonction de la taille et de la localisation peuvent avoir tendance à se fermer spontanément. D’autres ne se fermeront jamais ou auront même tendance à s’agrandir avec le temps.
Diagnostic
Le diagnostic de CIA peut se faire avant la naissance ("in utero") mais faire la différence avec un foramen ovale normalement présent avant la naissance n'est pas nécessairement facile. Il est capital qu’un médecin expérimenté en échocardiographie fœtale exclue des malformations cardiaques associées importantes, car ceci peut profondément changer la prise en charge et le pronostic. Si la CIA est isolée, il n’est pas nécessaire que l’enfant naisse dans un centre qui dispose d’une équipe cardiologique pédiatrique. Cependant, il est préférable que l’enfant soit examiné après quelques semaines de vie par un cardiologue pédiatre pour évaluer l’importance de cette CIA et guider les parents dans la prise en charge.
Dans la majorité des situations, le diagnostic est fait après la naissance. L’échocardiographie, permet à l’heure actuelle un diagnostic très complet de la cardiopathie et de ses conséquences. L’électrocardiogramme et la radiographie du thorax peuvent aider à évaluer l’importance de la CIA. Chez les adultes ou grands enfants, une échocardiographie transoesophagienne est parfois nécessaire pour bien visualiser la cloison entre les oreillettes, situé dans la partie postérieure du cœur.
Prise en charge
Un traitement par médicaments est en général non nécessaire pendant l’enfance car les patients sont peu symptomatiques. Les traitements médicaux sont par ailleurs peu efficaces chez les rares enfants symptomatiques.
Même en l’absence de symptômes, la fermeture de la CIA peut s’avérer nécessaire si la CIA engendre une augmentation significative de la quantité de sang allant vers le poumon et donc du travail cardiaque. Ceci est évalué en général par l’échocardiographie ou à la radiographie de thorax (dilatation du ventricule droit). Lorsque l’enfant est peu ou pas symptomatique (majorité des enfants), cette fermeture est en général proposée en âge préscolaire (5-6 ans). Dans les rares cas où l’enfant est très symptomatique dans les premières années, la fermeture peut être proposée avant. Lorsque le diagnostic de CIA est fait à l’âge adulte, le traitement par cathétérisme ou par chirurgie peut encore être proposé, même à un âge avancé, et ceci pour éviter les complications à long terme et ainsi augmenter l’espérance de vie.
Deux possibilités thérapeutiques existent pour la fermeture de CIA : le cathétérisme interventionnel ou la chirurgie.
Lorsque la CIA est bien ‘centrale’ dans la cloison interauriculaire (en termes médicaux le type ‘secundum’), et pas trop grande, la fermeture de la CIA peut être réalisée par cathétérisme cardiaque interventionnel. Ceci implique, sous anesthésie générale, l’introduction via les veines vers le cœur d’une prothèse qui fermera le trou. Cette technique qui permet d’éviter la chirurgie à cœur ouvert, est utilisée depuis plus de 15 ans et donne des résultats très satisfaisants. Plusieurs types de prothèse sont actuellement disponibles sur le marché.
La fermeture chirurgicale est la seule option pour certaines formes anatomiques de CIA’s situées près des valves ou des veines d’accès au cœur (CIA primum ou CIA type sinus venosus). De la même façon, si une CIA de type ‘secundum’ est trop large, ou l’enfant trop petit, il faut recourir à la fermeture chirurgicale. Il s’agit d’une chirurgie à cœur ouvert nécessitant une circulation extracorporelle (CEC) pour assurer la perfusion sanguine des différents organes, pendant que le chirurgien répare le cœur. Cette intervention, existant depuis plus de 50 ans, comporte actuellement très peu de risques (proche de 0%). Elle peut être pratiquée par sternotomie (cicatrice à l’avant) ou par thoracotomie (cicatrice en dessous du sein ou sur le côté).
Pour plus d'informations sur les traitements : voir le chapitre "TRAITEMENT des CARDIOPATHIES"
L’avenir des enfants opérés de CIA est en général excellent. La fermeture de la CIA corrige entièrement la malformation lorsque celle-ci est isolée. Un suivi cardiologique reste cependant nécessaire toute la vie, étant donné la présence de ‘cicatrices’ cardiaques. La pratique sportive est autorisée chez ces patients et les femmes peuvent en général envisager des grossesses.
 retour vers "Les communications anormales"
retour vers "Les communications anormales"
2. Communications entre les ventricules
 Description
Description
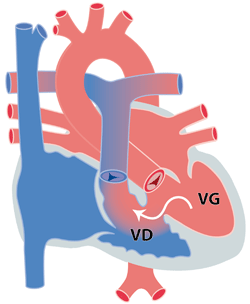
Le terme ‘communication interventriculaire’ ou CIV indique un passage existant entre le ventricule droit et gauche alors que dans un cœur ‘normal’ la paroi entre ces deux ventricules est tout à fait étanche et intacte (figure 1). Cette communication permet au sang rouge de passer du ventricule gauche (VG) vers le ventricule droit (VD) et ainsi vers les artères pulmonaires. Ceci entraîne une augmentation du travail du cœur et une augmentation de sang dans les poumons.
Il faut distinguer la CIV isolée et la CIV associée à d’autres malformations importantes. Ainsi, la CIV est obligatoire dans la ‘tétralogie de Fallot’ ou dans le ‘tronc artériel commun’ (voir plus loin). Elle peut également se voir en présence d’une coarctation de l’aorte, d’une interruption de l’arc aortique, et bien d’autres malformations cardiaques. Nous ne parlerons dans ce paragraphe que de la CIV isolée ou se présentant comme la malformation principale.
La CIV peut se localiser à différents endroits (différents types anatomiques) et il est très important de distinguer ces différentes localisations car la prise en charge médicale peut être très différente.
La CIV isolée représente à peu près 10% des malformations cardiaques congénitales.
Elle peut, bien que rarement lorsque la CIV est isolée, s’associer à d’autres malformations non cardiaques ou être associée à des anomalies chromosomiques comme la microdélétion 22q11, la trisomie 21 ou 18.
Présentation clinique
A la naissance, le nouveau-né avec une CIV isolée est en général cliniquement bien. En effet, les poumons sont fermés et ‘inutiles’ avant la naissance, et il faut plusieurs jours voire semaines après la naissance pour qu’ils s’ouvrent complètement et laissent circuler le sang facilement dans les vaisseaux (‘adaptation néonatale’). Peu de sang passe donc par la CIV vers les poumons dans les premiers jours de vie et l’enfant est asymptomatique. Dans les jours et semaines qui suivent la naissance, suite aux modifications pulmonaires, l'évolution dépendra essentiellement de la taille de la CIV. Si la CIV est large, le ventricule gauche, qui travaille à plus haute pression, envoie une grande quantité de sang par la CIV vers l’artère pulmonaire et donc les poumons. L’enfant présente alors progressivement un tableau de ‘défaillance cardiaque’ : essoufflement et endormissement lors de la prise des biberons, transpiration abondante, respiration rapide, même en dehors des repas. La prise de poids est souvent difficile. Un ‘souffle’ est audible à l'auscultation témoignant du passage en excès du sang vers les poumons. Le pédiatre peut observer une ‘polypnée’ (respiration rapide) ou une ‘tachycardie’ (battements du cœur rapides) ou même une ‘hépatomégalie’ (gros foie), tous témoins d’un travail cardiaque trop important. Lorsque la CIV est plus petite, l’enfant est souvent peu ou même pas du tout symptomatique. Parfois, les premiers mois de vie sont marqués par une respiration un peu plus rapide, une transpiration un peu plus abondante, une croissance un peu plus difficile ou uniquement des infections pulmonaires plus fréquentes et traînantes. Lorsque la CIV est très petite, les enfants peuvent ne présenter aucune plainte. Au plus la CIV est petite, au plus le souffle entendu par le médecin pourra être important. En effet, si le trou est petit, la différence de pression entre le ventricule gauche et le droit se maintient et le passage du sang d’une cavité à haute pression vers une cavité à basse pression fait beaucoup de bruit.
L’évolution clinique est variable en fonction de la taille mais également en fonction de la localisation de la CIV (type anatomique). En effet, certains types de CIV peuvent avoir tendance à se fermer spontanément alors que d’autres ne se fermeront jamais. Certaines CIV situées près des valves du cœur, peuvent avec le temps perturber le fonctionnement des valves, ce qui pourra changer la présentation clinique et nécessiter une intervention chirurgicale.
Diagnostic et prise en charge médicale
Le diagnostic de CIV peut se faire avant la naissance ("in utero"). Il est capital dans ces cas là qu’un médecin expérimenté en échocardiographie fœtale exclue des malformations cardiaques associées importantes, car ceci peut profondément changer la prise en charge et le pronostic. Si la CIV est isolée, il n’est pas nécessaire que l’enfant naisse dans un centre qui dispose d’une équipe cardiologique pédiatrique. Cependant, il est préférable que l’enfant soit examiné la première semaine de vie par un cardiologue pédiatre pour évaluer l’importance de cette CIV et guider les parents dans la prise en charge.
Dans la majorité des situations, le diagnostic est fait après la naissance. L’échocardiographie, permet à l’heure actuelle un diagnostic très complet de la cardiopathie et de ses conséquences. L’électrocardiogramme et la radiographie du thorax pourront aider à évaluer l’importance de la CIV. Il est de plus en plus rare qu’un cathétérisme cardiaque soit nécessaire pour mettre au point une CIV. Si d’autres anomalies non-cardiaques sont présentes, un bilan génétique à la recherche d’anomalies chromosomiques ou génétiques est indiqué.
Un traitement médical est en général nécessaire lorsque la CIV est de taille moyenne ou grande en présence de signes cliniques de défaillance cardiaque, signalés plus haut. Trois types de médicaments peuvent être utilisés :
1) les diurétiques (lasix®, dytenzide®) qui en favorisant la production d’urine, éliminent l’excédant d’eau dans le corps et surtout dans le poumons,
2) les inhibiteurs de l’enzyme de conversion (zestril®, renitec®), qui en abaissant la tension artérielle favorisent le passage du sang vers l’aorte plutôt que vers les artères pulmonaires et diminuent le travail du cœur.
Chez le nourrisson il faut souvent ajouter du fer afin d’éviter l’anémie qui engendre une moindre tolérance de la cardiopathie. L’alimentation est souvent rendue hypercalorique chez les nourrissons afin de favoriser la prise pondérale (concentration supérieure à la normale du lait 1er âge, rajout de sucre et parfois d’huile). En cas de gros problèmes alimentaires, une alimentation par gavage pourra être introduite, en milieu hospitalier.
3) la digoxine (lanoxin®), qui en améliorant la contractilité des fibres musculaires du cœur, aide le cœur face à un travail majoré. Son efficacité est toutefois actuellement discutée.
Chez le nourrisson il faut souvent ajouter du fer afin d’éviter l’anémie qui engendre une moindre tolérance de la cardiopathie. L’alimentation est souvent rendue hypercalorique chez les nourrissons afin de favoriser la prise pondérale (concentration supérieure à la normale du lait 1er âge, rajout de sucre et parfois d’huile). En cas de gros problèmes alimentaires, une alimentation par gavage pourra être introduite, en milieu hospitalier.
Une intervention chirurgicale sera nécessaire la première année de vie (de préférence avant 6 mois) lorsque la CIV reste large ou lorsque l’enfant tolère mal la CIV (mauvaise croissance, polypnée importante, décompensation cardiaque), et ceci quelle que soit sa localisation.
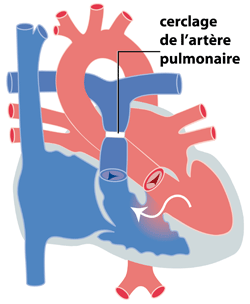
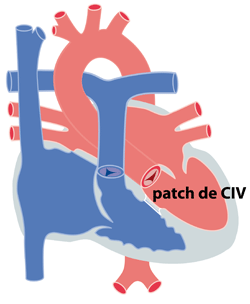
Lorsque la CIV est large, elle soumet en effet les poumons à de hautes pressions (équivalentes aux pressions dans l’aorte). Ceci peut endommager de façon définitive les poumons, si cet excès de pression se maintient pendant plus d’un an et engendrer ainsi une hypertension artérielle pulmonaire irréversible. Si l’enfant est jugé trop petit ou la CIV trop large pour une intervention visant une fermeture de l'orifice, un premier temps chirurgical consistera à mettre un anneau serrée autour de l’artère pulmonaire (cerclage ou banding) limitant par là la quantité de sang et la pression dans les poumons (figure 2a). La CIV pourra alors être fermée secondairement, lorsque l’enfant sera plus grand, dans de meilleures conditions. La plupart du temps cependant, le cerclage de l’artère pulmonaire ne sera pas nécessaire et la CIV pourra être fermé immédiatement. L’intervention est une intervention à cœur ouvert, nécessitant la mise en place d’une circulation extracorporelle (CEC) pour assurer la perfusion sanguine des différents organes, pendant que le chirurgien répare le cœur. De façon générale, le chirurgien utilise un patch en Dacron (tissu synthétique qui n’engendre aucun rejet) pour réaliser la fermeture de la CIV (figure 2b).
 |
 |
| (figure 2a) |
(figure 2b) |
Lorsque la CIV est plus petite une prise en charge interventionnelle est en général non nécessaire la première année de vie. Certaines CIV peuvent devenir tellement petites ou même se fermer complètement de façon à ne pas du tout nécessiter d'intervention. D’autres nécessiteront quand même une fermeture plus tardive, soit parce qu’elles maintiennent un niveau de travail cardiaque trop élevée, soit en raison de leur localisation ou de perturbation des valves adjacentes.
Même si la fermeture se fait encore souvent par voie chirurgicale, il existe depuis quelques années la possibilité pour certaines CIVs, de fermeture percutanée, c’est à dire, par cathétérisme cardiaque interventionnel, au moyen de prothèses introduites par les veines jusque dans le cœur. Les bonnes indications pour cette technique augmentent avec le temps suite à l'amélioration du matériel et de la technique elle-même.
Pour plus d'informations sur les traitements : voir le chapitre "TRAITEMENT des CARDIOPATHIES"
Les résultats de la fermeture chirurgicale ou par cathétérisme sont à l’heure actuelle très satisfaisants avec un risque très faible (< 2%) de complications graves . Les risques s’élèvent légèrement lorsque l’enfant doit être opéré très tôt dans la vie (premiers mois). L’avenir des enfants opérés de CIV est en général excellent. La fermeture de la CIV corrige la malformation. Un suivi cardiologique reste cependant nécessaire toute la vie, étant donné la présence de ‘cicatrices’ cardiaques. La pratique sportive est autorisée chez ces patients et les femmes peuvent en général envisager des grossesses.
retour vers "Les communications anormales"
3. Canal atrio-ventriculaire
 Description
Description
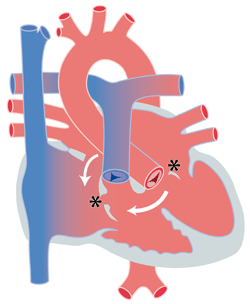
Le canal atrio-ventriculaire se caractérise pas une malformation des valves entre les oreillettes et les ventricules (valve auriculo-ventriculaire) et les parois adjacentes situées entre les oreillettes et les ventricules (figure). On distingue 3 formes : le canal atrio-ventriculaire complet (CAVC), le canal atrio-ventriculaire de type intermédiaire et la communication interauriculaire (CIA) de type primum avec fente mitrale.
Figure : schéma du canal atrio-ventriculaire. Les communications interventriculaire et interauriculaire permettent un passage de sang des cavités gauches vers les cavités droites (flèches). La malformation des valves atrio-ventriculaires engendre fréquemment des fuites valvulaires (astérisque)
Dans le canal atrio-ventriculaire complet (CAVC), la malformation est la plus importante. Au lieu d’avoir une valve mitrale et une valve tricuspide bien séparée, une seule grande valve existe, appelée valve atrio-ventriculaire. De part et d’autre de cette valve, la paroi séparant les oreillettes et les ventricules est partiellement déficiente. Il y a donc une communication interauriculaire (CIA) et une communication interventriculaire (CIV) associées.
A l’autre extrémité de la malformation, il y a la communication interauriculaire (CIA) primum avec fente mitrale. Les valves mitrale et tricuspide sont distinctes mais la valve mitrale est légèrement mal formée par la présence d’une fente, pouvant engendrer une fuite de cette valve plus ou moins importante. Il y a un déficit de la paroi entre les oreillettes associé, dans la portion juste à côté des valves mitrale et tricuspide. C’est ce que l’on appelle une CIA primum (par opposition à la CIA secundum, qui est la CIA situé au milieu de la paroi, voir plus haut).
Entre ces deux extrêmes, se situe le CAV de type intermédiaire. Les valves mitrale et tricuspide sont en général distinctes mais mal formées et on parle plutôt de valve auriculo-ventriculaire gauche et droite. Il y a en général une CIA et une CIV présente mais une des deux peut parfois manquer ou être très petite.
Le canal atrio-ventriculaire représente à peu près 7% des cardiopathies congénitales. Le canal atrio-ventriculaire est une malformation très fréquente chez l’enfant avec trisomie 21. Ainsi, en cas de CAVC, une trisomie 21 sera retrouvée dans presque 50% des cas.
Présentation clinique
La présentation clinique varie fortement en fonction de la forme.
En cas de canal atrio-ventriculaire complet, la symptomatologie est très semblable à celle d’une large CIV (voir chapitre des CIV). Après quelques jours ou semaines de vie, en raison de la baisse des résistances pulmonaires (‘adaptation néonatale’), l’enfant présente progressivement des difficultés alimentaires, une respiration rapide et une transpiration abondante surtout lors des repas avec une difficulté à prendre correctement du poids. En raison d’une CIA associé et la présence d’une valve atrio-ventriculaire unique qui est parfois fuyante, la symptomatologie peut même être plus marquée. Le médecin peut entendre un souffle et retrouver les différents signes témoins d’un tableau de ‘défaillance cardiaque.
En cas de CIA primum avec fente mitrale la symptomatologie se rapproche de celle de la CIA isolée (voir chapitre des CIA). Beaucoup d’enfants sont donc peu symptomatiques. Il est rare que la fente engendre une fuite sévère mais dans ce cas, les enfants peuvent être moins tolérants à l’effort et présenter un retard de croissance plus important. La présence d’un souffle cardiaque peut être le seul signe, amenant au diagnostic.
Les formes intermédiaires ont également une symptomatologie intermédiaire: en fonction de la taille de la CIV et de la CIA, en fonction de l’atteinte des valves, la symptomatologie sera plus ou moins importante.
Diagnostic et prise en charge
Le diagnostic du canal atrio-ventriculaire peut se faire avant la naissance. Il est capital comme pour les autres malformations cardiaques, qu’un médecin expérimenté en échocardiographie fœtale exclue des malformations cardiaques associées importantes, car ceci peut profondément changer la prise en charge et le pronostic. Il est également important de détecter les anomalies non cardiaques associées et en général, en présence d’un canal atrio-ventriculaire complet ou intermédiaire, une amniocentèse sera proposée afin de rechercher une trisomie 21. Si le canal atrio-ventriculaire est isolé, il n’est pas nécessaire que l’enfant naisse dans un centre qui dispose d’une équipe cardiologique pédiatrique. Cependant, il est préférable que l’enfant soit examiné la première semaine de vie, surtout en cas de canal atrio-ventriculaire complet et intermédiaire par un cardiologue pédiatre pour évaluer l’importance de la malformation et guider les parents dans la prise en charge.
En cas de diagnostic après la naissance, l’échocardiographie permet à l’heure actuelle une description très complète de la cardiopathie et de ses conséquences. L’électrocardiogramme et la radiographie du thorax font en général partie du bilan initial. Le cathétérisme cardiaque est parfois réalisé dans les formes intermédiaires pour mieux évaluer les conséquences pulmonaires. Si d’autres anomalies non-cardiaques sont présentes, un bilan génétique à la recherche d’anomalies chromosomiques (en particulier la trisomie 21) est réalisé.
Un traitement médical est en général nécessaire dans le cas de canal atrio-ventriculaire complet car le passage de sang du cœur gauche vers les poumons via les communications et donc le travail cardiaque est très augmenté. Le traitement est le même qu’en cas de CIV isolée (voir chapitre CIV). Le traitement médical n’est en général par nécessaire en cas de CIA primum et fente mitrale. En cas de forme intermédiaire, le traitement médical pourra parfois s’avérer nécessaire.
Un traitement chirurgical est toujours nécessaire car cette malformation ne guérit jamais spontanément. Ces lésions ne peuvent pas être traitées par cathétérisme cardiaque en raison des lésions valvulaires associées. Le type et l’âge au moment de la réparation sont très dépendants de la forme et de la présentation clinique. En cas de canal atrio-ventriculaire complet, en raison de la large CIV, il faut intervenir dans les 6 premiers mois de vie afin d’éviter les lésions irréversibles au niveau du poumon (hypertension artérielle pulmonaire). En général, le chirurgien procède à une réparation complète dans laquelle il ferme les communications interauriculaires et interventriculaires et répare au mieux la valve. Ceci est la partie la plus délicate de l’intervention car le chirurgien doit créer 2 valves au départ d’une seule et essayer de rendre celles-ci les plus ‘fonctionnelles’ possibles, sans trop de régurgitation (fuite) ni d’obstacle à l’écoulement du sang (sténose). C'est une intervention à cœur ouvert, nécessitant la mise en place pendant l’intervention, d’une ‘circulation extracorporelle’ pour assurer la perfusion sanguine des différents organes, pendant que le chirurgien répare le cœur. Si cependant l’enfant est trop petit ou que la malformation parait peu propice à une bonne réparation au vu du poids de l’enfant, le chirurgien peut d’abord procéder à un cerclage de l’artère pulmonaire (‘banding’, voir chapitre des CIV), ce qui permet de protéger les poumons. La chirurgie correctrice peut être réalisée plus tardivement dans de meilleures conditions, lorsque l’enfant sera plus grand. En cas de CIA primum avec fente mitrale, le traitement chirurgical est le plus souvent postposé jusque l’âge de 3-4 ans. Dans le cas des formes intermédiaires, le moment du traitement dépend surtout de la taille de la CIV. Si celle-ci est importante, l’enfant est opéré dans les premiers mois de vie, comme dans la forme complète. Si la CIV est petite, l’intervention peut se faire plus tardivement.
Pour plus d'informations sur les traitements : voir le chapitre "TRAITEMENT des CARDIOPATHIES"
Les résultats de la fermeture chirurgicale sont à l’heure actuelle très satisfaisants avec un risque faible de complications graves faible. Les risques s’élèvent cependant légèrement lorsque l’enfant doit être opéré très tôt dans la vie (premiers mois). Le résultat à long terme dépend surtout des lésions valvulaires et de la possibilité qu’a eue le chirurgien de ‘réparer’ les valves. Plus ou moins 10% des enfants nécessitent une réintervention au niveau de la valve mitrale à un certain moment de leur vie.
L’avenir des ces enfants opérés est en général très bon au niveau cardiaque mais sera quand même dépendant de la présence ou non de lésions valvulaires (surtout mitrale) résiduelles. Un suivi cardiologique reste nécessaire toute la vie, surtout pour le suivi de la fonction des valves. La pratique sportive est en général autorisée, en absence de lésions résiduelles significatives. Les femmes désireuses d’avoir un enfant, peuvent en général, en l’absence de lésions valvulaires résiduelles importantes, envisager une grossesse.
retour vers "Les communications anormales"
4. Communications entre les grands vaisseaux : canal artériel, fistules et collatérales
 Description
Description
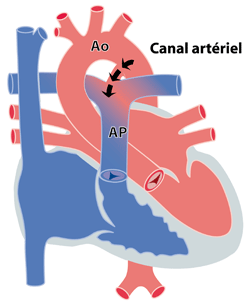
Les communications entre les grands vaisseaux c'est-à-dire l’aorte et les artères pulmonaires, sont de plusieurs types. Le plus fréquent est la persistance du canal artériel (CA). Le canal artériel, petit vaisseau situé entre l’aorte descendante et l’artère pulmonaire gauche est indispensable au bon fonctionnement du cœur avant la naissance. Normalement, il se ferme dans les premiers jours suivant la naissance. Parfois cependant, ce canal ne se ferme pas spontanément. Ceci engendre le passage de sang rouge oxygéné de l’aorte (Ao) via le canal artériel vers les artères pulmonaires (AP), entraînant un excès de sang vers les poumons et une augmentation du travail cardiaque (figure). Cette anomalie est assez fréquente surtout chez les enfants nés prématurément.
Il faut distinguer le CA isolé et le CA associé à d’autres malformations importantes. Ainsi, par exemple, la présence du CA est obligatoire dans certaines malformations comme l’atrésie pulmonaire ou l’interruption de l’aorte aortique, où sa fermeture spontanée entraîne une forte dégradation de l’état de l’enfant évoluant vers le décès si le canal n’est pas réouvert (voir plus loin). Nous ne parlerons dans ce chapitre que du canal artériel isolé ou se présentant comme la malformation principale.
D’autres communications anormales peuvent exister entre les gros vaisseaux, comme la fenêtre aorto-pulmonaire qui est une communication, ou plutôt un manque de cloisonnement entre l’aorte ascendante et l’artère pulmonaire centrale. Cette malformation est beaucoup plus rare que le canal artériel persistant. Certaines malformations cardiaques complexes et cyanogènes peuvent s’accompagner de collatérales aorto-pulmonaires, petits vaisseaux anormaux reliant l’aorte et les vaisseaux pulmonaires. Ces collatérales peuvent également exister, mais plus rarement, en l’absence d’autres malformations cardiaques. Comme le canal artériel persistant, ces deux types de communications augmentent l’arrivée de sang aux poumons et le travail cardiaque.
Présentation clinique
A la naissance, l’enfant ayant une communication anormale entre les grands vaisseaux est en général cliniquement bien. Dans les jours suivant la naissance, la diminution des ‘résistances’ pulmonaires (voir plus haut) favorise le passage anormal de sang par le CA ou la fenêtre aorto-pulmonaire. C’est donc quelques jours après la naissance que les symptômes peuvent apparaître mais ceci dépend essentiellement de la taille de la communication. Si la communication est large, l’enfant présente rapidement des signes de ‘défaillance cardiaque’: respiration rapide, transpiration lors des repas, difficultés pour finir les biberons, difficultés de prise pondérale. Chez l’enfant prématuré avec large canal artériel, les symptômes peuvent survenir très rapidement et être très sévères, nécessitant une prise en charge rapide. Le pédiatre ausculte un ‘souffle’ témoignant du passage en excès du sang vers les poumons, constate une ‘polypnée’ (respiration rapide) ou une ‘tachycardie’ (battements du cœur rapides), tous témoins d’un travail cardiaque trop important. Lorsque la communication est petite l’enfant est souvent peu ou pas du tout symptomatique et le diagnostic est souvent posé par la présence d’un souffle. Parfois une transpiration excessive à l'effort, une croissance un peu difficile, des infections pulmonaires plus fréquentes et traînantes peuvent être présents.
L’évolution clinique est variable. Le canal artériel et les collatérales aorto-pulmonaires peuvent spontanément se fermer avec dans ce cas disparition progressive des symptômes. Les fenêtres aorto-pulmonaires par contre ne se ferment jamais.
Diagnostic et prise en charge médicale
Le diagnostic de CA persistant ne peut pas se faire avant la naissance car il est normal pendant la grossesse que le canal artériel soit largement ouvert. La fenêtre aorto-pulmonaire peut être visualisée par l’échocardiographie fœtale bien que ceci puisse s’avérer difficile.
Le diagnostic des communications entre les gros vaisseaux est donc en général fait après la naissance. L’échocardiographie permet à l’heure actuelle un diagnostic très complet de la cardiopathie et de ses conséquences. L’électrocardiogramme et la radiographie du thorax pourront aider à évaluer l’importance de la communication. Il est de plus en plus rare qu’un cathétérisme cardiaque soit nécessaire pour le diagnostic.
Les traitements médicamenteux à base de diurétiques, digoxine et/ou inhibiteurs de l’enzyme de conversion (voir traitement de la CIV) peuvent améliorer les signes de défaillance cardiaque induits par les larges communications. Chez l’enfant prématuré avec canal artériel persistant large, l’administration par perfusion intraveineuse des certains médicaments (ibuprofène, indométacine) peut favoriser la fermeture du canal artériel. Malheureusement, ces médicaments sont en général inefficaces chez l’enfant né à terme avec canal artériel persistant.
Lorsque la communication reste large, une intervention est nécessaire, préférentiellement avant 6 mois de vie pour éviter les conséquences néfastes et irréversibles au niveau des poumons. L’importance de la symptomatologie nécessite par ailleurs souvent une intervention plus précoce. En cas de canal artériel, l’intervention peut être chirurgicale ou par cathétérisme, essentiellement en fonction du poids de l’enfant et la forme du canal artériel. L’intervention chirurgicale qui consiste à ligaturer le canal artériel n’est pas une intervention à cœur ouvert et se réalise via une incision thoracique latérale gauche. De plus en plus souvent, grâce à l’évolution de techniques, la fermeture du canal par cathétérisme cardiaque interventionnel peut être proposée. Dans ce cas, une petite prothèse est introduite via les veines ou artères de l’enfant jusqu’au cœur de l’enfant, afin de boucher le canal. Ce traitement évite l’ouverture du thorax et donc la cicatrice et permet un retour à domicile rapide après l’intervention. En cas de fenêtre aorto-pulmonaire, le traitement est chirurgical. Il s’agit d’une chirurgie à cœur ouvert, nécessitant une assistance circulatoire pendant l’intervention.
Lorsque la communication est moins large et n’engendre pas d’excès de pressions au niveau des poumons, le traitement peut être réalisé plus tard (souvent après l’âge d’un an). Pour le canal artériel et les collatérales aorto-pulmonaires, la fermeture se fait par cathétérisme cardiaque interventionnel. Certaines formes de petites fenêtres aorto-pulmonaires peuvent également bénéficier de cette technique, les autres seront traités par chirurgie.
Pour plus d'informations sur les traitements : voir le chapitre "TRAITEMENT des CARDIOPATHIES"
Les résultats de la fermeture par chirurgie ou par cathétérisme sont très bons avec un risque très faible (< 2%) de complications graves. Les risques s’élèvent cependant légèrement lorsque l’enfant est opéré très tôt dans la vie (enfant prématuré ou jeune nourrisson). L’avenir des ces enfants est en général excellent. L’intervention corrige entièrement la malformation. Un suivi cardiologique occasionnel reste cependant conseillé, étant donné la présence de ‘cicatrices’ cardiaques. La pratique sportive est autorisée et les femmes peuvent en général envisager des grossesses.
retour vers "Les communications anormales"



