2. Les anomalies des valves et vaisseaux
A. Les rétrécissements pulmonaires

Description
Les rétrécissements pulmonaires se définissent par la présence d’un obstacle (sténose) au passage du sang entre le ventricule droit et les artères pulmonaires. Cet obstacle peut se situer à différents niveaux:
1. en dessous de la valve pulmonaire : sténose sous-valvulaire pulmonaire
2.
au niveau de la valve pulmonaire : sténose valvulaire pulmonaire ou atrésie valvulaire pulmonaire
3.
au dessus de la valve pulmonaire : sténose supra-valvulaire pulmonaire
4.
au niveau des artères pulmonaires : sténose unilatérale ou bilatérale (gauche et droite) des artères pulmonaires
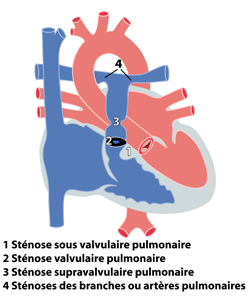
L’obstacle peut être localisé à plusieurs endroits chez un même patient. Les rétrécissements pulmonaires font souvent partie à part entière de malformations cardiaques complexes (tétralogie de Fallot, ventricule unique …). Nous parlerons dans ce chapitre uniquement des rétrécissements pulmonaires isolés. Ils peuvent également être associés à d’autres malformations non cardiaques et faire partie de ‘syndromes’ comme par exemple le syndrome de Williams , le syndrome de Noonan, le syndrome d’Alagille etc.
La sténose valvulaire pulmonaire est de loin la plus fréquente. Elle représente 7 % des malformations cardiaques congénitales. La valve pulmonaire est normalement très fine et formé de 3 feuillets (‘cusps’) s’ouvrant et se séparant complètement lors de l’éjection de sang à travers la valve (systole). Lors de la fermeture de la valve (diastole), les 3 feuillets s’apposent parfaitement assurant ainsi une étanchéité quasi parfaite de la valve. En cas de sténose, la valve peut être soit très épaisse, mal formée ou avoir simplement les feuillets attachés l’un à l’autre, empêchant ainsi une bonne ouverture de la valve en systole. Une fuite anormale de la valve en diastole peut y être associée.
En cas d’atrésie valvulaire pulmonaire, beaucoup plus rare, il n’y a aucun passage possible par la valve. La circulation pulmonaire dépend donc du canal artériel. Pendant la vie fœtale, cette absence d’ouverture de la valve pulmonaire empêche le ventricule droit de grandir normalement. Les enfants avec atrésie valvulaire pulmonaire isolée ont donc en général un ventricule droit plus petit que la normale (ventricule droit ‘hypoplasique’). Cette hypoplasie du ventricule droit est cependant très variable d’un enfant à l’autre. Dans les cas sévères, le ventricule droit est insuffisant pour assurer la circulation pulmonaire avec donc une situation de ‘ventricule unique’. (voir plus loin)
Les sténoses sous-valvulaires et supra-valvulaires pulmonaires sont rarement isolées. En général une sténose valvulaire y est associée ou une autre malformation cardiaque. En dessous de la valve, l’obstacle peut être musculaire ou fibreux ou une combinaison des deux. L’apparition de cet obstacle peut être favorisée par une communication interventriculaire associée, parfois même déjà fermée. Au dessus de la valve, il s’agit soit d’un réel manque de développement de l’artère pulmonaire centrale (hypoplasie) soit éventuellement d’une membrane obstruant partiellement le passage.
Les sténoses isolées des artères pulmonaires sont plus fréquentes à gauche qu’à droite en raison de l’attache du canal artériel de ce côté. Les sténoses bilatérales des artères pulmonaires s’inscrivent souvent dans des syndromes comme le syndrome de Williams, le syndrome d’Alagille etc.
Présentation clinique
En cas de sténose valvulaire pulmonaire très serrée (‘critique’), ou d’atrésie valvulaire pulmonaire l’enfant est symptomatique dans les premières heures voir jours de vie. En effet, lors de la fermeture spontanée du canal artériel, le débit sanguin pulmonaire est mis en péril. L’enfant devient très cyanosé. En l’absence de traitement rapide approprié ceci peut entraîner le décès de l’enfant. En cas de sténose valvulaire un souffle cardiaque typique est présent, absent par contre en cas d’atrésie.
La plupart des enfants avec sténose valvulaire serrée sont heureusement moins malades. Un passage ‘suffisant’ de sang vers les poumons est possible, même après fermeture du canal artériel. L’enfant peut cependant être discrètement cyanosé, mais c’est en général la présence d’un souffle à l’auscultation qui amène au diagnostic. L’obstacle valvulaire entraîne un excès de travail du ventricule droit ce qui en l’absence de traitement approprié peut amener à des signes de fatigue du ventricule droit (fatigue, essoufflement à l’effort, gonflement des jambes) pouvant mettre la vie du patient en danger.
En présence d'un obstacle modéré, qu’il soit valvulaire ou autre, les enfants sont en général pas ou peu symptomatiques et le seul signe d’appel est un souffle cardiaque à l’auscultation. Il en va de même pour les sténoses unilatérales des artères pulmonaires. Une sténose sévère d’une artère peut néanmoins compromettre la bonne croissance du poumon le rendant moins fonctionnel et plus sujet aux infections. En cas de sténoses sévères des 2 artères pulmonaires (bilatérales), le travail du ventricule droit est majoré, pouvant amener, en cas de lésions sévères à des signes de fatigue du ventricule droit.
Diagnostic et prise en charge médicale
Le diagnostic d’atrésie pulmonaire et de sténose valvulaire pulmonaire peuvent se faire avant la naissance. Il est capital dans ces cas là qu’un médecin expérimenté en échocardiographie fœtale exclue des malformations cardiaques associées importantes, car ceci peut profondément changer la prise en charge et le pronostic. En cas d’atrésie valvulaire ou de sténose valvulaire serrée il est nécessaire que l’enfant naisse dans un centre qui dispose d’une équipe cardiologique pédiatrique. En cas de sténose valvulaire peu serrée, l’enfant peut éventuellement naître ‘ailleurs’. Cependant, il est préférable que l’enfant soit examiné les premières semaines de vie par un cardiologue pédiatre pour évaluer l’importance de cette sténose pulmonaire et guider les parents dans la prise en charge.
Liens
En cas de suspicion après la naissance d’atrésie valvulaire pulmonaire ou de sténose valvulaire pulmonaire serrée, il est urgent que l’enfant soit rapidement transféré dans un centre qui dispose d’une équipe cardiologique pédiatrique. En cas de cyanose importante, le médecin peut avant le transfert débuter un traitement par prostaglandine E (prostin®) en intraveineux, afin de rouvrir le canal artériel et ainsi améliorer l’oxygénation de l’enfant.
Le diagnostic des obstacles pulmonaires est posé par échocardiographie. Un électrocardiogramme et une radiographie de thorax font en général partie du bilan initial. En cas de sténose des artères pulmonaires, une résonance magnétique, ou un scanner thoracique ou éventuellement un cathétérisme cardiaque s'avèrent souvent utiles pour mieux définir la lésion, surtout chez l'enfant plus grand. En cas de malformations non cardiaques associées, un bilan complémentaire génétique est généralement réalisé.
Le traitement des enfants avec sténose valvulaire pulmonaire sévère se fait par cathétérisme cardiaque interventionnel. Des sondes à ballonnets sont introduits via la veine fémorale (pli de l’aine) jusqu’au cœur. Le gonflement du ballon au niveau de la valve pulmonaire permettant une meilleure ouverture de la valve dans la majorité des cas et présente peu de risque. La plupart des enfants sont ‘guéris’ après la procédure et ne nécessitent plus de traitement ultérieur.
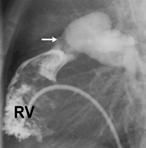 |
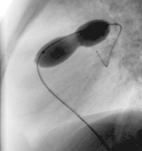 |
La figure montre un exemple de sténose valvulaire pulmonaire traitée par valvuloplastie percutanée. L’image de gauche montre l’injection de produit de contraste dans le ventricule droit (RV). Celle-ci permet de voir la valve pulmonaire (flèche blanche). Après mensuration, un ballon de diamètre adéquat est introduit, en suivant le même chemin, et gonflé à hauteur de la valve (image de droite), dégonflé puis retiré du cœur de l’enfant.
|
En cas d’atrésie valvulaire pulmonaire, un cathétérisme cardiaque est également effectué dans les premiers jours de vie. Le cathétérisme cardiaque permet avec l’échocardiographie, de décider si le ventricule droit est suffisamment développé pour assurer la circulation vers les poumons. Si le ventricule droit est trop petit, l’enfant est considéré comme ayant une ‘circulation univentriculaire’. La prise en charge en est détaillée ailleurs. Si le ventricule droit est estimé assez grand, une ouverture de cette valve est tentée souvent lors du même cathétérisme au moyen d’une sonde chauffante (‘radiofréquence’) qui brûle un trou dans la valve, trou qui est ensuite agrandi par des ballons de taille croissante. Lorsque ceci n’est pas possible, le chirurgien ouvre la valve par chirurgie à cœur ouvert. Certains enfants nécessitent ultérieurement d'autres procédures par cathétérisme ou par chirurgie afin d’ouvrir davantage la voie pulmonaire.
Les sténoses sévères des artères pulmonaires se traitent également par cathétérisme cardiaque. L’efficacité de la dilatation au ballon est cependant moindre qu’au niveau de la valve pulmonaire et il faudra souvent s’aider de stents pour maintenir les vaisseaux ouverts.
La chirurgie est réservé aux situations ou le cathétérisme cardiaque ne permet pas de lever l’obstacle mais également aux sténoses sous-valvulaires pulmonaires et supra-valvulaires pulmonaires qui ne sont pas traitables par cathétérisme.
Lorsque l’obstacle sur la voie pulmonaire est peu sévère, une simple surveillance est suffisante. Avec le temps les lésions peuvent s’aggraver mais parfois aussi diminuer voire disparaître. C’est en général l’échocardiographie qui décidera à quel moment un traitement s’avère nécessaire.
L’avenir des enfants traités pour rétrécissement pulmonaire dépend essentiellement de l’absence d'obstacle résiduel important. Ainsi, l’avenir des enfants avec sténose valvulaire pulmonaire isolée est très bon car la simple dilatation au ballon permet la plupart du temps de lever l’obstacle. Par contre, en cas de sténoses sévères bilatérales des artères pulmonaires ayant incomplètement répondu au traitement et évoluant défavorablement avec le temps, l’avenir peut être plus incertain.
Un suivi cardiologique est nécessaire toute la vie, quel que soit le résultat du traitement. En l’absence de lésion résiduelle significative, le sport est en général autorisé. En présence de lésion résiduelle ceci doit être discuté avec le cardiologue. Il en va de même en cas de désir de grossesse.
Pour plus d'informations sur les traitements : voir le chapitre "TRAITEMENT des CARDIOPATHIES"
 retour vers "Les anomalies valves et vaisseaux"
retour vers "Les anomalies valves et vaisseaux"
B. les rétrécissements et fuites aortiques
Description
Les rétrécissements aortiques se définissent par la présence d’un obstacle au passage du sang entre le ventricule gauche et l’aorte. Cet obstacle peut se situer à différents niveaux :
1. en dessous de la valve aortique : sténose sous-valvulaire aortique
2. au niveau de la valve aortique : sténose valvulaire aortique
3. au dessus de la valve aortique : sténose supra-valvulaire aortique
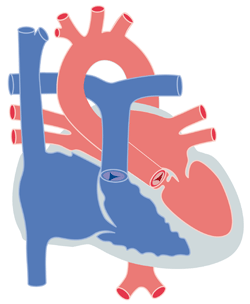
Figure 1
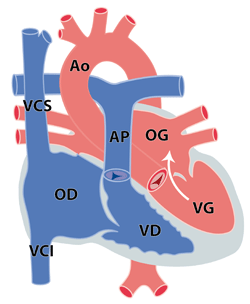
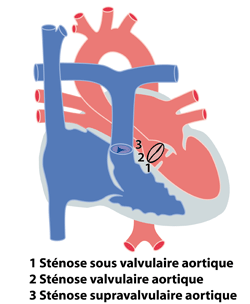 L’obstacle peut être localisé à plusieurs endroits chez un même patient (figure 1). Les rétrécissements aortiques sont fréquemment associés à une coarctation de l'aorte. Ils peuvent faire partie de cardiopathies plus complexes. Nous ne traiteront dans ce chapitre que les formes isolées.
L’obstacle peut être localisé à plusieurs endroits chez un même patient (figure 1). Les rétrécissements aortiques sont fréquemment associés à une coarctation de l'aorte. Ils peuvent faire partie de cardiopathies plus complexes. Nous ne traiteront dans ce chapitre que les formes isolées.
La sténose valvulaire aortique est la plus fréquente. Elle représente 3 % des malformations cardiaques congénitales. La valve aortique est normalement très fine et formé de 3 feuillets (tricuspide) s’ouvrant et se séparant complètement lors de l’éjection de sang à travers la valve (systole). Lors de la fermeture de la valve (diastole), les 3 feuillets s’apposent parfaitement assurant ainsi une étanchéité parfaite de la valve. En cas de sténose, la valve peut être soit très épaisse, mal formée ou avoir les feuillets attachés l’un à l’autre, empêchant ainsi une bonne ouverture de la valve en systole. Une valve aortique bicuspide (‘bicuspidie’), c'est-à-dire faite de 2 feuillets au lieu de 3, est par ailleurs présente chez 1% de la population. La bicuspidie est parfois ‘familiale’ ce qui veut dire que plusieurs membres d’une même famille peuvent en être porteur. Une sténose est plus fréquemment rencontrée en présence d’une valve bicuspide par rapport à la valve aortique tricuspide, soit dès la naissance soit à l'âge adulte.
Une valve anormale sténosée peut également mal se fermer en diastole, générant ainsi une fuite ou ‘régurgitation’ aortique. Cette fuite peut chez certains patients être plus importante que la sténose voir être isolée (sans sténose).
Les sténoses sous-valvulaires (figure 2) sont isolées ou associés à une sténose valvulaire aortique. L’obstacle est musculaire ou fibreux ou une combinaison des deux. Elles sont souvent absentes à la naissance et se développent avec l’âge. L’apparition d’une lésion sous-valvulaire peut être favorisée par l’existence d’une communication interventriculaire.
 |
| Figure 2 : sténose sous-valvulaire aortique |
Les lésions supravalvulaires se caractérisent pas un manque de développement (hypoplasie) de la région de l’aorte juste au dessus de l’émergence des coronaires. Ces formes sont typiques du syndrome de Williams. Elles peuvent également être présentes chez plusieurs membres de la famille, en dehors de tout syndrome (formes familiales, non syndromiques).
Présentation clinique
En cas de sténose valvulaire aortique très serrée (‘critique’), l’enfant peut être symptomatique dans les premières heures voir jours de vie. En effet, lors de la fermeture spontanée du canal artériel, le débit vers l’aorte est mis en péril. L’enfant se met rapidement en situation de défaillance cardiaque puis de choc cardiaque marqué par une pâleur, une respiration rapide, un refus de boire, les extrémités très froides. A l’examen clinique, le médecin ausculte un souffle cardiaque et des pouls très faibles. En l’absence de traitement rapide approprié ceci peut entraîner le décès de l’enfant.
La plupart des enfants, même avec sténose aortique serrée sont heureusement moins malade. Un passage ‘suffisant’ de sang vers l’aorte est possible, même après fermeture du canal artériel. C’est en général le souffle typique qui amène au diagnostic. L’obstacle aortique entraîne un excès de travail du ventricule gauche qui en absence de traitement approprié peut engendrer une fatigue du ventricule gauche ou des signes de mauvaise perfusion des artères coronaires situées en aval de l’obstacle. Ceci se manifeste par un essoufflement à l’effort, des douleurs précordiales à l’effort ou des malaises à l’effort pouvant être graves et mortels.
En présence d'un obstacle modéré, qu’il soit valvulaire ou autre, les enfants sont généralement pas ou peu symptomatiques et le seul signe d’appel est alors un souffle cardiaque à l’auscultation.
Diagnostic et prise en charge médicale
Le diagnostic de sténose aortique peut se faire avant la naissance. Il est capital dans ces cas là qu’un médecin expérimenté en échocardiographie fœtale exclue des malformations cardiaques associées importantes, car ceci peut profondément changer la prise en charge et le pronostic. En cas de sténose valvulaire serrée il est nécessaire que l’enfant naisse dans un centre qui dispose d’une équipe cardiologique pédiatrique. En cas de sténose valvulaire peu serrée, l’enfant peut éventuellement naître ‘ailleurs’. Cependant, il est préférable que l’enfant soit examiné les premières semaines de vie par un cardiologue pédiatre pour évaluer l’importance de cette sténose aortique et guider les parents dans la prise en charge.
En cas de suspicion après la naissance de sténose valvulaire aortique serrée, il est urgent que l’enfant soit rapidement transféré dans un centre qui dispose d’une équipe cardiologique pédiatrique. En cas de défaillance cardiaque ou choc cardiaque, le médecin peut avant le transfert débuter un traitement par prostaglandine E (prostin®) en intraveineux, afin de rouvrir le canal artériel et ainsi améliorer la circulation sanguine.
Le diagnostic est posé par échocardiographie. Un électrocardiogramme et une radiographie de thorax font en général partie du bilan initial. En cas de sténose supra-valvulaire aortique, un bilan complémentaire génétique ou familial peut être proposé.
Le traitement des enfants avec sténose valvulaire aortique sévère est possible par cathétérisme cardiaque interventionnel (dilatation de la valve par un ballonnet) ou par chirurgie, à cœur ouvert. Le choix sera dépendant essentiellement de l’âge du patient, de l’aspect de la valve mais aussi en fonction de l’expérience et l’habitude de l’équipe médico-chirurgicale. Par exemple, chez un jeune enfant avec une valve très rétrécie mais ne fuyant pas, la dilatation au ballon est une option valable. Par contre, chez un adolescent avec une sténose serrée mais également une fuite significative de la valve, on procède plutôt à une correction chirurgicale. Le chirurgien peut soit essayer de ‘réparer’ la valve, soit si ceci est impossible, la remplacer. Chez l’enfant, lorsqu’un remplacement valvulaire aortique est nécessaire, l’intervention choisie est en général l’intervention de ‘Ross’, d’après le chirurgien qui l’a décrit. Cette intervention utilise la valve pulmonaire du patient pour remplacer la valve aortique. Ensuite, une valve de donneur est mise en place de la valve pulmonaire. Cette intervention permet d’éviter l’utilisation d’une valve mécanique, nécessitant l’administration à vie d’un traitement anticoagulant que l’on préfère éviter chez l’enfant. Les sténoses supra-valvulaires et sous-valvulaires sont toujours traitées par chirurgie car ces lésions ne répondent pas à la dilatation par ballonnet.
Les sténoses modérées ne nécessitent en général pas de traitement mais une simple surveillance. Avec le temps les lésions peuvent devenir plus sévères et nécessiter alors un traitement. C’est en général l’échocardiographie qui détermine le moment où un traitement s’avère nécessaire.
Les résultats de ces différents traitements sont en général très satisfaisants même si les risques et succès dépendent grandement de l’âge de l’enfant, de la sévérité et de la morphologie de l’endroit rétrécie. L’avenir des enfants traités pour rétrécissement aortique dépend essentiellement de l’absence ou non de lésion résiduelle importante. Ainsi, en cas de sténose ou fuite valvulaire résiduelle significative, les risques de réintervention sont élevés. En cas de sténose sous-valvulaire aortique, même après résection chirurgicale complète, le risque de récidive existe.
Un suivi cardiologique est donc nécessaire toute la vie, dans tous les cas, quel que soit le résultat du traitement. En l’absence de lésion résiduelle significative, le sport sera en général autorisé. En présence de lésion résiduelle ceci devra être discuté avec le cardiologue. Il en va de même en cas de désir de grossesse.
Pour plus d'informations sur les traitements : voir le chapitre "TRAITEMENT des CARDIOPATHIES"
retour vers "Les anomalies valves et vaisseaux"
C. la coarctation et l’interruption de l’arc aortique
Description
Le terme ‘coarctation de l’aorte’ signifie un rétrécissement de l’aorte dans sa portion descendante. Ce rétrécissement gêne le passage du sang dans sa route vers la partie inférieure du corps.
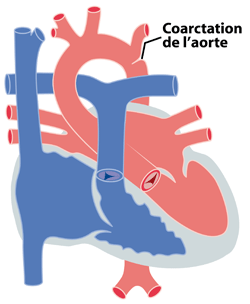
Figure 1 : en cas de coarctation localisée, le rétrécissement est juste après l ‘émergence des vaisseaux du cou.
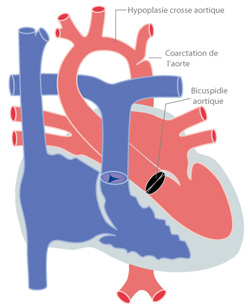
Figure 2 : coarctation associée à une hypoplasie de la crosse et à une bicuspide valvulaire aortique.
La coarctation peut se présenter comme un rétrécissement très localisé (figure 1), sans autres anomalies de l’aorte. Par contre elle peut s’associer à une hypoplasie (c'est-à-dire un manque de développement) de toute la crosse aortique (figure 2) qui est alors beaucoup plus petite que normalement. Cette hypoplasie contribue évidemment à l’obstacle.
Dans 50% des cas, la coarctation de l’aorte est associée à une bicuspidie de la valve aortique.
Une CIV est parfois associée à la coarctation (‘syndrome de coarctation’). La coarctation peut s’inscrire dans un contexte de malformation cardiaque plus complexe comme le ventricule unique, le canal atrio-ventriculaire complet, etc. Nous ne parlerons dans cet article que de la coarctation isolée ou se présentant comme la malformation principale.
La coarctation peut de façon très rare se localiser au niveau de l’aorte abdominale (‘coarctation abdominale’). Il s’agit à ce moment là souvent d’un long segment rétréci.
Elle peut, bien que rarement lorsque la coarctation est isolée, s’associer à d’autres malformations non cardiaques ou être associée à des anomalies chromosomiques comme le syndrome de Turner ou le syndrome de Williams. La coarctation représente près de 5% des malformations cardiaques congénitales
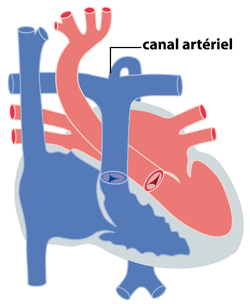 L’interruption de l’arc aortique, beaucoup plus rare, est en quelque sorte une forme extrême de coarctation. Il y a absence de continuité entre l’aorte ascendante et l’aorte descendante (figure 3). Elle est en général associée à une CIV ou parfois à des malformations plus complexes comme le tronc artériel ou le ventricule unique. Elle peut être associée à une anomalie génétique bien précise, la microdélétion 22q11.
L’interruption de l’arc aortique, beaucoup plus rare, est en quelque sorte une forme extrême de coarctation. Il y a absence de continuité entre l’aorte ascendante et l’aorte descendante (figure 3). Elle est en général associée à une CIV ou parfois à des malformations plus complexes comme le tronc artériel ou le ventricule unique. Elle peut être associée à une anomalie génétique bien précise, la microdélétion 22q11.
Figure 3 : interruption de l'arc aortique. Le canal artériel est indispensable pour la perfusion de l'aort descendante.
Présentation clinique
La présentation clinique de la coarctation varie fortement en fonction de l’âge, la forme anatomique et l’importance du rétrécissement.
Lorsque le rétrécissement est important ou en cas d’interruption de l’arc aortique, des symptômes très sévères peuvent apparaitre rapidement après la naissance. Tant que le canal artériel est ouvert, l’enfant présente peu de symptômes. La fermeture du canal quelques heures ou jours après la naissance rend très difficile le passage du sang vers la partie inférieure du corps (foie, reins, membres inférieurs..) et le travail cardiaque est alors fortement augmenté. L'enfant évolue rapidement vers une défaillance cardiaque sévère puis un état de ‘choc’. L'enfant est pâle, respire vite et refuse les biberons. Les médecins constatent outre la pâleur et la respiration rapide, une absence de pouls fémoraux (dans le pli de l’aine) et une impossibilité de prendre la tension artérielle dans les jambes (car le sang y passe très mal). Une fois le diagnostic établi, une prise en charge rapide est indispensable.
La présentation est heureusement souvent moins dramatique, même chez le nouveau né. En effet, lorsque le rétrécissement est moins sévère, le sang continue à arriver à la moitié inférieure du corps, même après fermeture du canal artériel. Le cœur doit cependant travailler plus fort pour ‘vaincre’ l’obstacle. Les parents et médecins peuvent voire apparaitre des petits signes de défaillance cardiaque (respiration rapide surtout à l’effort) mais c’est en général la découverte d’une absence de pouls fémoraux, un souffle cardiaque ou une tension artérielle dans le bras beaucoup plus élevé que dans la jambe qui amène au diagnostic.
Chez le grand enfant et l’adulte, c’est essentiellement une hypertension artérielle, lorsque la tension est prise au bras ou un souffle qui amènent au diagnostic. En cas de diagnostic tardif, des céphalées, douleurs thoraciques ou crampes dans jambes peuvent également être présents.
Diagnostic et prise en charge médicale
Le diagnostic de coarctation reste difficile avant la naissance en raison du fait que le canal artériel est largement ouvert pendant la vie fœtale, ce qui peut masquer la coarctation. Toutefois, un développement un peu ralenti du ventricule gauche (asymétrie des ventricules) ou une dilatation du ventricule droit peuvent être de signes évocateurs de développement néonatal de coarctation et peuvent justifier une surveillance rapprochée pendant les premiers jours de vie.
Lorsqu’un diagnostic de coarctation ou d’interruption de l’arc aortique est suspecté dans les premiers jours de vie, la prise en charge médicale peut être une urgence médicale. L’enfant doit être transféré dans un centre de cardiologie pédiatrique. En cas de signes de défaillance cardiaque, un traitement par perfusion de prostaglandines E (prostin ®) peut être administré afin de rouvrir le canal artériel et stabiliser l‘état clinique de l’enfant.
Chez le jeune enfant, l'échocardiographie permet le diagnostic de coarctation. Cet examen permet également d’exclure d’autres lésions associées et de vérifier le degré de défaillance du cœur (réduction de contractilité du cœur gauche). En cas d’interruption de la crosse aortique, un cathétérisme cardiaque est parfois nécessaire afin de décrire la morphologie de l’aorte et des vaisseaux en sortant
Chez le plus grand enfant avec coarctation, une résonance magnétique ou un scanner thoracique permet une meilleure visualisation de l’anatomie de l’aorte.
Le traitement chirurgical reste la meilleure option thérapeutique pour la coarctation chez le nouveau-né et le nourrisson. Le chirurgien excise le segment rétréci et élargit la crosse de l’aorte si nécessaire. Cette intervention se fait par incision sur le côté gauche du thorax, sans assistance circulatoire. Parfois néanmoins, lorsque la crosse de l’aorte est très petite et la réparation plus difficile, le chirurgien ouvre le thorax par devant et s’aide d’une assistance circulatoire pendant l’intervention. En cas d’interruption de l’arc aortique la reconstruction de la continuité de l’aorte nécessite également une assistance circulatoire pendant l’intervention.
Chez l’enfant plus grand avec coarctation, deux options existent: la dilatation par cathétérisme cardiaque interventionnel au moyen d’un ballon ou d’un stent (petit treillis métallique qui restera en place et maintiendra l’aorte bien ‘ouverte’) ou l’intervention chirurgicale. Le choix d’une intervention par rapport à l’autre dépend de l’âge, de la forme anatomique et de l’expérience du centre traitant l’enfant. Ainsi la dilatation au ballon est une bonne option en cas de lésion très localisée, sans hypoplasie de la crosse de l’aorte et pas trop serrée. Chez un adulte avec une forme assez serrée mais bien localisé, la mise en place d’un stent est souvent la meilleure option.
Les deux types d’interventions présentent des risques et complications, heureusement peu fréquentes (<5%), la plupart pouvant être traitées sans séquelles à long terme. Une récidive de la coarctation après traitement chirurgical ou par cathétérisme est possible (recoarctation) mais dépend fortement de l’âge à l’intervention, la morphologie initiale de l’aorte et la technique qui a pu être utilisée. La recoarctation est plus fréquente après chirurgie d’interruption de l’arc aortique. La recoarctation peut le plus souvent être traitée efficacement par dilatation au ballon ou par stent(cathétérisme cardiaque). Lorsque le diagnostic et le traitement d’une coarctation sont réalisés tardivement, le patient peut garder un certain degré d’hypertension artérielle, nécessitant parfois un traitement par médicaments. De façon générale, un patient ayant eu une coarctation est plus à risque de souffrir d’hypertension à l’âge adulte. Des contrôles occasionnels de la tension artérielle sont donc nécessaires. Comme toute cardiopathie opérée, un suivi cardiologique régulier reste nécessaire. Le sport est en général autorisé, en absence de recoarctation ou lésion résiduelle significative tout en évitant néanmoins les sports violents et les sports à un niveau de compétition. Les grossesses peuvent être envisagées en l’absence de recoarctation ou de lésions résiduelles significatives mais un suivi régulier pendant la grossesse peut s’avérer nécessaire.
Pour plus d'informations sur les traitements : voir le chapitre "TRAITEMENT des CARDIOPATHIES"
retour vers "Les anomalies valves et vaisseaux"
D. les anomalies de la valve mitrale
Description
Les anomalies isolées de la valve mitrale sont rares. Elles sont par contre plus fréquemment associées à d’autres malformations comme par exemple l’hypoplasie du cœur gauche, le ventricule droit à double issue, la sténose aortique etc.
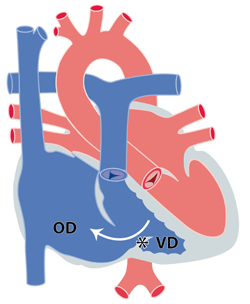
 On distingue les malformations responsables d’une fuite ou régurgitation anormale de la valve mitrale et celles responsables d’une mauvaise ouverture et donc d’une sténose mitrale. Certaines lésions peuvent engendrer une sténose et régurgitation.
On distingue les malformations responsables d’une fuite ou régurgitation anormale de la valve mitrale et celles responsables d’une mauvaise ouverture et donc d’une sténose mitrale. Certaines lésions peuvent engendrer une sténose et régurgitation.
Les malformations les plus fréquentes responsables de régurgitation sont les fentes mitrales, le prolapsus mitral et la valve mitrale à double orifice. En cas de fente mitrale isolée, un des feuillets de la valve mitrale est réellement ‘fendu’, ce qui génère une fuite lors de la contraction du cœur (‘systole’)(figure 1). Le prolapsus mitral, rare chez l’enfant est par contre fréquent chez l’adulte, surtout chez la femme. Il peut être retrouvé en cas de maladie de Marfan et autres maladies affectant les tissus conjonctifs. En cas de prolapsus, un des feuillets de la valve mitrale est trop grand et possède des piliers (attaches à la paroi du ventricule) trop longs. Ceci empêche une fermeture correcte de la valve, générant donc une fuite. La valve mitrale à double orifice est une malformation rare, où la valve possède un deuxième orifice qui ne se ferme pas en systole, et engendre donc une fuite.
Figure 1 : fente mitrale générant une fuite lors de la systole (flèche)
 La valve mitrale peut être diffusément trop petite (valve hypoplasique) ou présenter des anomalies des piliers et cordages attachant la valve au ventricule (valve en ‘parachute’ ou en ‘hamac’) (figure 2). Ces différentes anomalies empêchent l’ouverture complète de la valve et un bon passage du sang de l’oreillette vers le ventricule (sténose). Il peut également exister juste au dessus de la valve mitrale une membrane, appelée membrane pré-mitrale, qui si elle est très serrée peut empêcher le passage du sang et donc engendrer une sténose.
La valve mitrale peut être diffusément trop petite (valve hypoplasique) ou présenter des anomalies des piliers et cordages attachant la valve au ventricule (valve en ‘parachute’ ou en ‘hamac’) (figure 2). Ces différentes anomalies empêchent l’ouverture complète de la valve et un bon passage du sang de l’oreillette vers le ventricule (sténose). Il peut également exister juste au dessus de la valve mitrale une membrane, appelée membrane pré-mitrale, qui si elle est très serrée peut empêcher le passage du sang et donc engendrer une sténose.
Figure 2 : valve mitrale en parachute avec anomalie des piliers de la valve.
Présentation clinique
Les anomalies de la valve mitrale peuvent se présenter à tout âge en fonction de la sévérité et de l’évolution de l’atteinte. Il est cependant exceptionnel d’avoir des enfants très symptomatique dans les premiers jours de vie.
En cas de régurgitation mitrale, l’enfant est rarement symptomatique tant que la fuite n’est pas importante. C’est donc souvent la présence d’un souffle qui permet de poser le diagnostic. En cas de fuite importante, l’enfant présente une mauvaise tolérance à l’effort avec essoufflement. La régurgitation peut également être associée à des troubles du rythme cardiaque, ressenti comme des palpitations. Des signes de défaillance cardiaques peuvent apparaitre si une fuite sévère est non traitée.
En cas de sténose mitrale, le mauvais passage du sang est responsable d’une stagnation de sang dans les poumons (œdème pulmonaire). Ceci engendre un essoufflement à l’effort et chez le jeune enfant une respiration rapide (polypnée). Le souffle est souvent peu audible et c’est donc rarement par cette voie que le diagnostic est posé. En cas de sténose sévère, la stagnation du sang dans les poumons engendre un excès de travail pour le cœur droit, ce qui peut mener à la défaillance du cœur droit (intolérance à l’effort, rétention d’eau dans les jambes ou le ventre, gros foie…)
Diagnostic et prise en charge médicale
Les anomalies mitrales peuvent en théorie être décelées par échocardiographie fœtale bien que ceci peut s’avérer difficile en absence de fuite ou de sténose sévère. Comme pour les autres malformations, il est capital qu’un médecin expérimenté en échocardiographie fœtale exclue, dans la mesure du possible, des malformations cardiaques associées importantes, car ceci peut profondément changer la prise en charge et le pronostic.
Le diagnostic est souvent fait après la naissance, par échocardiographie. Un électrocardiogramme et parfois une radiographie de thorax font en général partie du bilan initial. Chez le grand enfant, l’échocardiographie transoesophagienne, permet souvent de mieux visualiser la morphologie exacte de la valve.
Une lésion peu sévère ne nécessite aucun traitement hormis une surveillance régulière car elle peut évoluer avec le temps. En cas de régurgitation significative visualisée à l’échocardiographie, les médicaments visant à alléger le travail du cœur sont utiles. Ces médicaments appartiennent à la classe des inhibiteurs de l’enzyme de conversion (captopril (capoten®), enalapril (renitec®), lisinopril (zestril®) …) parfois associés à des diurétiques (furosémide (lasix®), …). En cas de sténose symptomatique, avec ‘œdème pulmonaire’, des diurétiques (furosémide (lasix®), …) peuvent ‘tempérer’ la situation. Si la fuite ou la sténose sont sévères ou mal tolérées une intervention chirurgicale s’impose. Le chirurgien ‘répare’ la valve (valvuloplastie) ou enlève la membrane pré-mitrale responsable de l’obstacle. Dans les rares cas ou la réparation n’est pas possible, un remplacement de la valve est effectué, en général par une valve mécanique. Ces deux interventions sont des interventions à cœur ouvert.
Un suivi cardiologique est nécessaire toute la vie, dans tous les cas, quel que soit le résultat du traitement. L’avenir de ces enfants est fonction de la sévérité de la lésion et de la présence ou non de lésions résiduelles après chirurgie éventuelle. La pratique de sport est en général autorisée mais doit être discutée avec le cardiologue. En cas de sténose sévère ou fuite sévère, le sport est déconseillé en attendant la chirurgie. De même, en cas de désir de grossesse chez les patientes à l’âge adulte, ceci doit être discuté avec le médecin responsable.
Pour plus d'informations sur les traitements : voir le chapitre "TRAITEMENT des CARDIOPATHIES"
retour vers "Les anomalies valves et vaisseaux"
E. les anomalies de la valve tricuspide
Description
Les anomalies isolées de la valve tricuspide sont rares. Elles sont plus fréquemment associées à d’autres malformations comme par exemple l’atrésie de la valve pulmonaire.
On distingue les malformations responsables d’une fuite ou régurgitation anormale de la valve tricuspide et celles responsables d’une mauvaise ouverture et donc d’une sténose de la tricuspide. Certaines lésions peuvent engendrer sténose et régurgitation associées.
 La malformation la plus fréquemment responsable de régurgitation est la maladie d’Ebstein d’après le médecin ayant décrit la maladie. Dans cette anomalie, un des feuillets de la valve tricuspide est trop ‘bas’ inséré sur la paroi, ce qui empêche la bonne fermeture de la valve lors de la contraction du cœur (systole) (figure). La sévérité de cette maladie est très variable, essentiellement en fonction de l’importance de ce ‘déplacement’ du feuillet de la valve. Si le déplacement est très important, la maladie peut se présenter dès la naissance, sous une forme très sévère. Si le déplacement est peu sévère, le patient est généralement peu symptomatique. La moitié des patients avec maladie d’Ebstein ont une communication interauriculaire associée (CIA). La maladie d’Ebstein peut être associée à d’autres malformations plus complexes.
La malformation la plus fréquemment responsable de régurgitation est la maladie d’Ebstein d’après le médecin ayant décrit la maladie. Dans cette anomalie, un des feuillets de la valve tricuspide est trop ‘bas’ inséré sur la paroi, ce qui empêche la bonne fermeture de la valve lors de la contraction du cœur (systole) (figure). La sévérité de cette maladie est très variable, essentiellement en fonction de l’importance de ce ‘déplacement’ du feuillet de la valve. Si le déplacement est très important, la maladie peut se présenter dès la naissance, sous une forme très sévère. Si le déplacement est peu sévère, le patient est généralement peu symptomatique. La moitié des patients avec maladie d’Ebstein ont une communication interauriculaire associée (CIA). La maladie d’Ebstein peut être associée à d’autres malformations plus complexes.
Figure : maladie d'Ebstein caractérisée par une anomalie d'implantation d'un des feuillets de la valve triscupide, responsable d'une régurgitation (flèche)
Les autres malformations de la valve tricuspide, sont regroupées sous le terme de dysplasies de la valve tricuspide, pouvant être responsable d’une fuite ou d’une sténose tricuspide, ou les 2 associées.
Présentation clinique
Les anomalies de la valve tricuspide peuvent se présenter à tout âge en fonction de la sévérité et de l’évolution. Les cas sévères de maladies d’Ebstein se présentent dès la naissance. En effet, la fuite tricuspide importante, empêche le sang d’être éjecté vers les artères pulmonaires. Il passe donc essentiellement via le foramen ovale ou la CIA vers l’oreillette gauche et ensuite l’aorte. L’enfant est donc très bleu (cyanose). Cette cyanose peut s’accentuer lors de la fermeture spontanée du canal artériel à quelques heures ou jours de vie, mettant la vie de l’enfant en danger.
La plupart des enfants avec maladie d’Ebstein ou dysplasie tricuspide ont une présentation clinique moins sévère. Un essoufflement à l’effort est souvent le premier signe. Un souffle est audible. La régurgitation peut également être associée à des troubles du rythme cardiaque, ressenti comme des palpitations.
Diagnostic et prise en charge médicale
Les anomalies tricuspides, en particulier la maladie d’Ebstein peuvent en théorie être décelées par échocardiographie fœtale bien que ceci peut s’avérer difficile en absence de fuite sévère. Comme pour les autres malformations, il est capital qu’un médecin expérimenté en échocardiographie fœtale exclue, dans la mesure du possible, des malformations cardiaques associées importantes, car ceci peut profondément changer la prise en charge et le pronostic. Si un diagnostic de maladie d’Ebstein avec fuite est posé, il est conseillé que l’enfant naisse dans un centre disposant d’une équipe de cardiologie pédiatrique, afin que l’enfant puisse est pris en charge rapidement après la naissance.
Le diagnostic après la naissance se fait par échocardiographie. Un électrocardiogramme et parfois une radiographie de thorax font en général partie du bilan initial.
En cas de présentation néonatale de maladie d’Ebstein, la présence d’une cyanose sévère justifie l’administration intraveineuse de prostaglandine E (prostin®). Ce médicament permet de maintenir ouvert ou de réouvrir le canal artériel et d’ainsi améliorer l’arrivée de sang dans les artères pulmonaires. Ce médicament peut souvent être arrêté après quelques semaines. En effet, juste après la naissance, les poumons sont peu ouverts et les résistances pulmonaires élevées. Le sang s’écoule donc difficilement du ventricule droit vers les poumons, surtout en présence d’une fuite tricuspide importante. Progressivement, au cours des premières semaines de vie, les poumons vont s’ouvrir et les résistances pulmonaires baisser, permettant une meilleure arrivée de sang à partir du ventricule. Le canal artériel n’est alors donc plus nécessaire pour la perfusion des poumons. Lorsque l’arrêt de ce médicament s’avère impossible, la chirurgie en période néonatale est nécessaire.
La plupart des patients avec maladie d’Ebstein ou dysplasie tricuspide échappent à cette nécessité d’intervention néonatale. Le ‘timing’ de l’intervention est alors fonction du degré de fuite ou sténose évalué sur base des symptômes, de l’échocardiographie et d’autres examens comme l’épreuve d’effort, la résonance magnétique etc …Certains patients ne doivent jamais être opérés! Lors de l’intervention chirurgicale, le chirurgien ‘répare’ la valve (valvuloplastie). Pour la maladie d’Ebstein, ceci consiste à ‘rehausser’ le feuillet trop bas implanté. Un remplacement de la valve est rarement nécessaire.
Un suivi cardiologique est nécessaire toute la vie, dans tous les cas, quel que soit le résultat du traitement. L’avenir de ces enfants est fonction de la sévérité de la lésion et de la présence ou non de lésions résiduelles après chirurgie éventuelle. La pratique de sport est en général autorisée mais doit être discutée avec le cardiologue. En cas de sténose sévère ou fuite sévère, le sport est déconseillé en attendant la chirurgie. De même, en cas de désir de grossesse chez les patientes à l’âge adulte, ceci doit être discuté avec le médecin responsable.
Pour plus d'informations sur les traitements : voir le chapitre "TRAITEMENT des CARDIOPATHIES"
retour vers "Les anomalies valves et vaisseaux"
F. les lésions étagées
 DESCRIPTION-PRESENTATION CLINIQUE - DIAGNOSTIC ET PRISE EN CHARGE
DESCRIPTION-PRESENTATION CLINIQUE - DIAGNOSTIC ET PRISE EN CHARGE
Il est bien connu en cardiologie pédiatrique qu’une ‘malformation cardiaque peut en cacher une autre’. Cependant certaines malformations s’associent davantage entre elles et pas avec d’autres. Ainsi, les différentes malformations concernant le cœur gauche s’associent volontiers. Il en va de même, bien qu’en moindre mesure pour le cœur droit.
Cœur gauche
Comme signalé plus haut, la coarctation de l’aorte s’associe volontiers à une bicuspidie avec ou sans sténose de la valve aortique. En cas de sténose valvulaire aortique, on peut y voir une sténose sous-valvulaire aortique et même une sténose ou malformation de la valve mitrale.
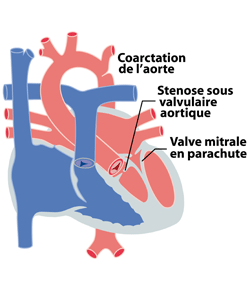 Ainsi, le syndrome de Shone, d’après le médecin l’ayant décrit, consiste en l’association d’une anomalie mitrale, aortique et d’une coarctation. Le syndrome de l’hypoplasie du cœur gauche (voir chapitre des ‘cœurs univentriculaires’) peut être considéré comme l’extrême: toutes les composantes du cœur gauche se sont mal développés y compris le ventricule gauche lui-même.
Ainsi, le syndrome de Shone, d’après le médecin l’ayant décrit, consiste en l’association d’une anomalie mitrale, aortique et d’une coarctation. Le syndrome de l’hypoplasie du cœur gauche (voir chapitre des ‘cœurs univentriculaires’) peut être considéré comme l’extrême: toutes les composantes du cœur gauche se sont mal développés y compris le ventricule gauche lui-même.
Plusieurs explications sont possibles pour expliquer ces associations : l’une génétique, l’autre lié au développement du cœur pendant la vie utérine.
En ce qui concerne la cause génétique, on sait que de multiples gènes (situés sur nos chromosomes) sont responsables du bon développement du cœur. On peut spéculer que certains gènes interviennent plus particulièrement dans le contrôle de la formation du cœur gauche. Des anomalies des ces gènes peuvent être responsables de malformations. En fonction de l’atteinte de ces gènes ou en fonction de facteurs environnants non précisées à ce jour, l’atteinte peut être plus ou moins sévère et atteindre plusieurs parties du cœur gauche. Les gènes, y compris les gènes malades, peuvent se transmettre à la descendance. Il n’est pas rare de voir des familles ou plusieurs membres de la famille présentent une pathologie du cœur gauche. Cependant, elle peut être différente d’un membre à l’autre. Ainsi dans la famille d’un enfant avec hypoplasie du cœur gauche, on peut parfois retrouver des parents, grands-parents ou famille plus éloignée avec bicuspidie de la valve aortique ou coarctation. Malheureusement, à ce jour, aucun gène responsable de la ‘pathologie’ gauche n’a encore pu être identifié mais les recherches continuent. Retrouver un gène responsable est comme retrouver une aiguille dans une botte de foin !
Les gènes ne sont pas les seuls responsables de l’étendu des malformations. Il y a également un côté ‘dynamique’ à la croissance du cœur. En effet, une malformation d’une valve du cœur gauche peut entrainer une mauvaise circulation du sang dans le cœur gauche pendant la vie utérine. Cette mauvaise circulation empêche le bon développement du reste du cœur gauche. Ainsi par exemple, l’existence d’une sténose aortique serrée peut favoriser l’apparition d’une coarctation de l’aorte avec hypoplasie de la crosse par manque de perfusion pendant la vie utérine.
La présentation clinique de ces associations de malformations est évidemment très variable, en fonction de la localisation et du degré des différentes lésions. La prise en charge et le pronostic peut également être très différents de ceux d’une lésion ‘isolée’. Chaque patient mérite donc une information très personnalisée.
Cœur droit
Les lésions du cœur droit peuvent également s’associer. Comme signalé plus haut, les rétrécissements pulmonaires peuvent se localiser à plusieurs endroits. Des malformations de la valve tricuspide peuvent être associées à des sténoses ou plus volontiers à des atrésies pulmonaires. Comme pour le cœur gauche, l’extrême est représenté par l’association d'une atrésie tricuspide avec une atrésie pulmonaire où le ventricule droit sera nécessairement mal développé (hypoplasie du cœur droit).
Comme pour le cœur gauche, les raisons de ces associations sont probablement génétiques mais également dynamiques.
La présentation clinique est dominée par la malformation la plus sévère. La prise en charge et le pronostic doivent être individualisés en fonction des lésions.
Pour plus d'informations sur les traitements : voir le chapitre "TRAITEMENT des CARDIOPATHIES"
retour vers "Les anomalies valves et vaisseaux"



